Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
| Code | CSB-EP018238HU |
| Abbreviation | Recombinant Human PMM2 protein |
| MSDS | |
| Size | $224 |
| Order now | |
| Image |
|
| Have Questions? | Leave a Message or Start an on-line Chat |
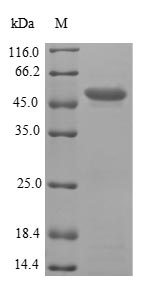
This Human PMM2 recombinant protein was produced in E.coli, where the gene sequence encoding Human PMM2 (1-246aa) was expressed with the N-terminal GST tag. The purity of this PMM2 protein was greater than 90% by SDS-PAGE.
One of the primary functions of PMM2 is to catalyze the isomerization reaction between mannose-6-phosphate and mannose-1-phosphate. This reaction is a crucial step in the mannose metabolism pathway, generating mannose-1-phosphate, which is utilized in the synthesis of various important glycoproteins and sugar molecules. PMM2 mediates the synthesis and metabolism of mannose, which is vital for multiple biological processes. Glycoproteins are a class of proteins with attached sugar molecules, and they play roles in cell signaling, cell adhesion, immune system function, and more. Mutations in PMM2 are associated with a genetic disorder known as Phosphomannomutase 2 deficiency (PMM2-CDG). This is a rare metabolic disorder that results in abnormal glycoprotein synthesis, affecting the function of various organs and systems, including the nervous system, muscular system, immune system, and others. The symptoms and severity of PMM2-CDG can vary among individuals.
There are currently no reviews for this product.

