Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
1. What is Autophagy?
Autophagy is a highly conserved adaptive process through which eukaryotic cells deliver dispensable or potentially dangerous cytoplasmic material to lysosomes for degradation under various conditions of cellular stress, including deprivation, growth factor depletion, infection, low energy and hypoxia. The main function of autophagy is to provide nutrients for vital cellular functions during fasting and other forms of stress, and is crucial to the maintenance of organismal homeostasis in both physiological and pathological situations[1][2].
2. The types of Autophagy
Thus far, there are three main types of autophagy, macroautophagy, microautophagy and Chaperone mediated autophagy, which are mediated by the autophagy-related genes and their associated enzymes[3][4][5]. Macroautophagy (referred to throughout this article as autophagy), is the main pathway which is primarily used to eradicate damaged cell organelles or unused proteins and divided into canonical and non-canonical autophagy. In this article, we focus on the research progress of macroautophagy.
a. Canonical Autophagy
Canonical autophagy, also considered as a nonselective process, is the highly conserved process by which eukaryotic cells scavenge their own cytoplasmic contents by sequestration into a phagophore and then fusion with a lysosome for degradation.
mTOR is the key regulator of the molecular mechanisms of canonical autophagy. autophagy-inducing stimus (such as nutrient deprivation, stress, hypoxia) triggers the activation of AMPK, whose kinase activity inhibits mTOR, and then activates the pre-initiation complex, composed of ULK1/2, ATG13, ATG101 and FIP200. This complex then activates the PI3K complex(VPS34, Beclin 1 and ATG14). The PI3K complex produces phosphatidylinositol 3-phosphate (PI3P), which acts as recruitment messenger for the downstream two ubiquitin-like conjugation systems. One of ubiquitin-like conjugation systems is responsible for the formation of a supramolecular protein complex containing ATG5, ATG12 and autophagy-related 16-like 1 (ATG16L1), including autophagy-related 7 (ATG7) and ATG10. another composes of ATG3, ATG4 and ATG7, promotes the cleavage of members of the Atg8 protein family, including human LC3, and their conjugation to phosphatidylethanolamine (PE). The activity and coordination of these two systems facilitate the expansion and sealing of the autophagosome, as well as the lipidation and embedding of LC3-PE into the autophagosomal membrane[6].
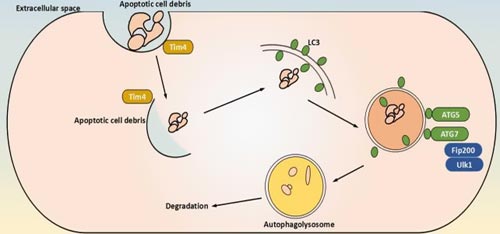
b. Non-canonical Autophagy
On the contrary, non-canonical autophagy is considered a specific process that selectively targets to internal cellular substrates. The selective autophagy is the autophagy of oragenelles, such as ribophagy[7], pexophagy[8], lipophagy[9], chlorophagy[10], ]mitophagy[11] and others.
Currently, mitophagy is a representive of selective autophagy, which can be regulated by serveral different mechanisms depending on the physiological context. Here are two different menchanisms to regulate mitophagy.
One key regulator is Parkin. Upon damage or depolarization, the mitochondrial kinase PteN-induced kinase 1 (PiNK1) becomes stabilized and recruits the Ub E3 protein ligase Parkin. PiNK1 and Parkin assemble phosphorylated Ub (pUb) chains on several proteins of the outer mitochondrial membrane via a feedforward mechanism. These proteins of the outer mitochondrial membrane in turn recruit cargo receptors such as calcium-binding and coiled-coil domaincontaining protein 2 (NDP52) and optineurin (OPtN). In this process, free Ub is phosphorylated by PiNK1, and Parkin attaches polyUb to the mitochondrial surface and the ubiquitin-like (uBl) domain of Parkin. these phosphorylation events enhance both the ubiquitin ligase activity of Parkin and its retention time on damaged mitochondria.

Another player in mitophagy is taNKbinding kinase 1 (tBK1), which promotes coupling of the cargo to the phagophore via phosphorylating ub-binding domains and LIRs of several cargo receptors, thereby increasing their affinity for pub and LC3, respectively. Notably, mitophagy can also occur in a ub-independent manner via mitochondrial proteins such as BCL2/adenovirus E1B protein-interacting protein 3-like (NiX), FuN14 domain-containing protein 1 (FuNDC1) and BCL2/adenovirus E1B protein-interacting protein 3 (BNiP3), which possess an LC3-interacting region (LIR) and therefore function as direct cargo receptors. they are typically regulated by stress-dependent phosphorylation. Finally, lipids, including phospholipids, such as cardiolipin and ceramide, have been shown to mediate mitophagy. in neuronal cells, cardiolipin is located at the inner membrane of healthy mitochondria, but upon mitochondrial damage, it is externalized and presented on the mitochondrial surface, where it is recognized by LC3[12][13].
3. Core autophagy proteins of the autophagic pathway
Autophagy is a cellular catabolic pathway involving in protein degradation, organelle turnover, and non-selective breakdown of cytoplasmic components. This progress is consist of six stages, including induction, phagophore nucleation, phagophore expansion, autophagosome formation, lysosome fusion, and component degradation. The core proteins of that stages are shown in the table 1 in details.
Table 1. Key autophagic factors and their regulation
| Protein | Function | Mechanisms of regulation |
|---|---|---|
| Initiation and phagophore nucleation | ||
| ULK1 and ATG1 | Serine/threonine kinase; initiates autophagy by phosphorylating components of the autophagy machinery | Stress and nutrients (via mTORC1, AMPK and LKB1); TFEB and several miRNAs |
| FIP200 | Component of ULK complex (possibly scaffolding function) | ULK1 and miRNAs |
| ATG13 | Adaptor mediating the interaction between ULK1 and FIP200; enhances ULK1 kinase activity | ULK1, mTORC1 and AMPK |
| ATG101 | Component of ULK complex; recruitment of downstream ATG proteins | ULK1 |
| VPS34 | Catalytic component of PI3KC3–C1; generates PI3P in the phagophore and stabilizes the ULK complex | AMPK, ULK1 and p300 (acetylation) |
| Beclin 1 | Promotes formation of PI3KC3–C1 and regulates the lipid kinase VPS34 | Activation: AMPK, ULK1, MAPKAPK2, MAPKAPK3, DAPK and UVRAG; inhibition: BCL-2, AKT and EGFR |
| ATG14 | PI3KC3–C1 targeting to the PAS and expanding phagophore | PIPKIγI5 and mTORC1 |
| ATG9 | Delivery of membrane material to the phagophore | ULK1 complex |
| WIPI2 | PI3P-binding protein that recruits ATG12-ATG5-ATG16L to the phagophore; retrieval of ATG9 from early autophagosomal membranes | TFEB (positive transcription regulator) and ZKSCAN3 (negative transcription regulator) |
| Phagophore expansion | ||
| ATG4 | Cysteine protease that processes pro-ATG8s; also, deconjugation of lipidated LC3 and ATG8s | ULK1 and ROS |
| ATG7 | E1-like enzyme; activation of ATG8; conjugation of ATG12 to ATG5 | miRNAs |
| ATG3 | E2-like enzyme; conjugation of activated ATG8s to membranal PE | miRNAs |
| ATG10 | E2-like enzyme that conjugates ATG12 to ATG5 | miRNAs |
| ATG12~ATG5–ATG16L | E3-like complex that couples ATG8s to PE | CSNK2 |
| PE-conjugated ATG8s | Scaffold for assembly of the ULK1 complex; supports membrane tethering and hemifusion events for phagophore expansion | ULK1, PKA, ATG4 and mTOR |
| ATG9 | Delivery of membrane material to the phagophore | ULK1 |
| Autophagosome formation | ||
| Ubiquitin | Cargo labelling | PINK (phosphorylation) |
| Cardiolipin and ceramide | Cargo labelling | Phosphorylation |
| p62 | Autophagy receptor | ULK1 and TBK1 |
| OPTN | Autophagy receptor | TBK1 |
| NBR1 | Autophagy receptor | TBK1 |
| NDP52 | Autophagy receptor | TBK1 |
| PE-conjugated LC3 | Interaction with autophagy receptors; also phagophore expansion and sealing | ULK1, PKA, ATG4 and mTOR |
| LC3s and GABARAPs | Unclear | Unclear; might involve phosphorylation and acetylation events |
| ATG4 | Removal of ATG8s from the surface of the autophagosome | Unknown |
| PE-conjugated LC3s and GABARAPs | Linking the autophagosome to microtubulebased kinesin motor | Unclear; might involve phosphorylation and acetylation events |
| Fusion with the lysosome | ||
| PE-conjugated LC3s and GABARAPs | Mediates autophagosome–lysosome fusion upon phosphorylation through PLEKHM1 and HOPS | STK3 and STK4 |
| ATG14 | Promotes SNARE-driven membrane fusion | Unknown |
| Rab GTPase RAB7 | Unclear | Unknown |
*Note: this content of the table 1 is driven from Ivan Dikic[14]
4. Autophagy and Disease
The field of autophagy research has developed rapidly since the first description of the process in the 1960s and the identification of autophagy genes in the 1990s. Autophagy is now increasingly studied at the level of organismal pathophysiology and is being connected to the medical sciences.
a. Autophagy and cancer
Autophagy is an important process during cancer progression, but the exact roles of autophagy are strongly context-dependent in cancer cells. It is thought that autophagy prevents cancer development[15]. In theory, high autophagic activity is believed to be cytoprotective and to suppress cancer initiation. However, once cancer is established, increased autophagic flux often enables tumour cell survival and growth[16][17]. In pre-malignant lesions, much evidence suggests that enhancers of autophagy might prevent cancer development[18]. Conversely, in advanced cancers, both enhancing autophagy and inhibiting it have been suggested as therapeutic strategies[19][20]. Thus, there is an important question in cancer therapy: should we try to enhance autophagy or should we try to inhibit it? In another word, autophagy has opposing, context-dependent roles in cancer, and interventions to both stimulate and inhibit autophagy have been proposed as cancer therapies.
b. Autophagy in inflammation and immunity
Numerous studies reveal that autophagy is involved in a variety of immune functions, such as inflammatory cytokine secretion, lymphocyte development, control of inflammation, antigen presentation[21][22][23][24] and removal of intracellular bacteria[25][26][27]. Emerging evidences demonstrate that autophagy play a crucial role in these functions through the susceptibility of autophagy-deficient animals[28][29].
Mechanistically, autophagy extensively crosstalks with inflammatory signalling cascades, including multiple context-specific and bidirectional interactions with the IKK–NF-κB pathway. Autophagy is induced by NF-κB via transactivating Beclin 1. Moreover, in the presence of various physiological and pharmacological stress stimuli, the IKK complex can induce autophagy[28]. However, the NF-κB pathway may also inhibit autophagy, for instance, in the context of tumour necrosis factor-α (TNFα)-induced cell death and in macrophages infected by Escherichia coli. Furthermore, in several cell lines, TNFα-driven NF-κB activation requires a functional autophagy pathway. Autophagy can also suppress NF-kB signalling by the autophagic degradation of active IKKβ, mediated either by KEAP1 (Kelch-like ECH-associated protein 1) or by the E3 ubiquitin-protein ligase RO52 (also known as TRIM21)[30][31][32].
References
[1] Noda, N. N. & Inagaki, F. Mechanisms of autophagy. Annu[J]. Rev. Biophys. 2015, 44, 101-122.
[2] Lorenzo Galluzzi1, José Manuel Bravo-San Pedro, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles[J]. Nat Rev Drug Discov. 2017 16(7):487-511.
[3] Mizushima N, Yoshimori T, et al. The role of Atg proteins in autophagosome formation[J]. Annual Review of Cell and Developmental Biology. 2011, 27 (1): 107-32.
[4] Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations[J]. Nature Cell Biology. 2007, 9 (10): 1102-9.
[5] Mizushima N, Ohsumi Y, et al. Autophagosome formation in mammalian cells[J]. Cell Structure and Function. 2002, 27 (6): 421-9.
[6] Payel Sil, Ginger Muse, et al. A ravenous defense: canonical and non-canonical autophagy in immunity[J]. Current Opinion in Immunology. 2018, 50:21-31.
[7] Ding, Wen-Xing; Yin, et al. Mitophagy: mechanisms, pathophysiological roles, and analysis[J]. Biological Chemistry. 2012, 393 (7).
[8] Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy[J]. Cell Death and Differentiation. 2013, 20 (1): 3–11.
[9] Till, Andreas, Lakhani, Ronak, et al. Pexophagy: The Selective Degradation of Peroxisomes[J]. International Journal of Cell Biology. 2012: 1–18.
[10] Lei, Lei. Chlorophagy: Preventing sunburn[J]. Nature Plants. 2017, 3 (3).
[11] An, Heeseon, Harper, J. Wade. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy[J]. Nature Cell Biology. 2018, 20 (2): 135–143.
[12] Chu, C. T. et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells[J]. Nat. Cell Biol. 2013,15, 1197–1205.
[13] Sentelle, R. D. et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy[J]. Nat. Chem. Biol. 2012, 8, 831–838.
[14] Ivan Dikic, and Zvulun Elazar. Mechanism and medical implications of mammalian autophagy[J]. Nat. Rev. Mol. Cell Biol. 2018.
[15] Jean M. Mulcahy Levy, Christina G. Towers, et al. Targeting autophagy in cancer[J]. Nat Rev Cancer. 2017, 17(9):528-542.
[16] Amaravadi, R., Kimmelman, et al. Recent insights into the function of autophagy in cancer[J]. Genes Dev. 2016, 30, 1913–1930.
[17] White, E. Deconvoluting the context-dependent role for autophagy in cancer[J]. Nat. Rev. Cancer. 2012, 12, 401–410 .
[18] Galluzzi, L. Autophagy in malignant transformation and cancer progression[J]. EMBO J. 2015.
[19] Levy, J. M. & Thorburn, A. Targeting autophagy during cancer therapy to improve clinical outcomes[J]. Pharmacol. Ther. 2011, 131, 130–141.
[20] Towers, C. G. & Thorburn, A. Therapeutic targeting of autophagy[J]. EBioMedicine. 2016, 14, 15–23.
[21] Thurston, T. L., Wandel, M. P., et al. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion[J]. Nature. 2012, 482, 414–418.
[22] Gomes, L. C. & Dikic, I. Autophagy in antimicrobial immunity[J]. Mol. Cell 2014, 54, 224–233.
[23] Wild, P. et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth[J]. Science. 2011, 333, 228–233.
[24] Saitoh, T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production[J]. Nature. 2008, 456, 264–268.
[25] Paludan, C. et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy[J]. Science. 2005, 307, 593–596.
[26] Loi, M. et al. Macroautophagy proteins control MHC Class I levels on dendritic cells and shape anti-viral CD8(+) T cell responses[J]. Cell Rep. 2016, 15, 1076–1087.
[27] Wei, J. et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis[J]. Nat. Immunol. 2016, 17, 277–285.
[28] Rockel, J. S. & Kapoor, M. Autophagy: controlling cell fate in rheumatic diseases[J]. Nat. Rev. Rheumatol. 2016, 12,517–531.
[29] Ma, Y., Galluzzi, L., Zitvogel, L, et al. Autophagy and cellular immune responses[J]. Immunity. 2013, 39, 211–227 .
[30] Copetti, T., Bertoli, C., et al. p65/RelA modulates BECN1 transcription and autophagy[J]. Mol. Cell. Biol. 2009, 29, 2594–2608.
[31] Schlottmann, S. et al. Prolonged classical NF-kappaB activation prevents autophagy upon E. coli stimulation in vitro: a potential resolving mechanism of inflammation[J]. Mediators Inflamm. 2008, 725854.
[32] Criollo, A. et al. Autophagy is required for the activation of NFkappaB[J]. Cell Cycle. 2012, 11, 194–199.
[33] Kim, J. E. et al. Suppression of NF-kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation[J]. Cell. Signal. 2012, 22, 1645–1654.yt


Comments
Leave a Comment