Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
In 1980, Nüsslein-Volhard et al. discovered a gene that determines the dorsal-ventral differentiation in the development of Drosophila embryos and named it Toll [1] [2]. Later, similar homologous receptors were found in mammals and were called Toll-like receptors (TLRs) [3].
TLRs are important receptors involved in innate immunity, and they can specifically identify and combine pathogen-associated molecular patterns (PAMPs), triggering a series of signal transduction, which leads to the release of inflammatory mediators, eventually activates the adaptive immune system [4] [12].
Among TLRs family, TLR4 (Toll-like receptor 4) is the most special member, which recognizes PAMPs both bacteria and viruses. And TLR4 is the only one that can utilize four adaptor molecules MyD88 (myeloid differentiation Primary-response protein 88), MAL (MyD88-adaptor-like protein, also called TIRAP), TRIF (TIR-domain-containing adaptor protein-inducing IFNB) and TRAM (TRIF-related adaptor molecule) to transmit cascade reaction, promote the secretion of inflammatory factors and interferon.
Bruce A. Beutler' group demonstrated that TLR4 is an important sensor for LPS by using the C3H/HeJ mouse strain which is known to have a hypoergia to LPS [5]. TLR4 can recognize LPS and thereby release a series of cytokines that promote proinflammatory response [27]. TLR4 is also known as an amplifier for the inflammatory reaction [6] [7].
TLR4 is widely distributed on the cell membranes of lymphocytes, epithelial cells, monocyte-macrophages, dendritic cells (DC), cardiomyocytes, etc. It belongs to type I transmembrane protein, which is composed of three regions: extracellular domain, transmembrane domain, intracellular domain. The extracellular domain is a leucine-rich repeat (LRR) that binds to CD14 and mediates the recognition of PAMPs. The intracellular domain is a highly conserved sequence, which is homologous with interleukin 1 receptors (IL-1R), so it is also known as TIR (Toll/IL-1 receptor) region [16]. After the TLR4 combined with corresponding ligands, signal transduction to TIR area, and then further activation of NFκB (nuclear factor κB) signaling pathway and MAPK (mitogen-activated protein kinase) signaling pathway, so as to promote the activation of a variety of inflammatory cytokine gene expression [8].
First, LPS binding protein (LBP) binds to LPS to form an LPS/LBP complex, which is recognized and bound by CD14. Alone, CD14 cannot transmit signals within cells because it lacks a transmembrane domain. Instead, it transfers LPS into hydrophobic pockets within the MD-2 (myeloid differentiation 2). MD-2 is a secreted glycoprotein belonging to the MD-2-associated lipid recognition family. Although there is no transmembrane domain, MD-2 can be attached to the cell surface through its interaction with TLR4 through specific epitopes. Upon binding of LPS, a receptor multimer consisting of two copies of the TLR4-MD-2-LPS complex is formed [13] [14] [15].
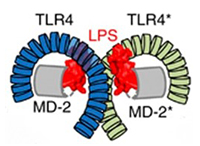
Figure 1 LPS recognition by TLR4–MD-2
(sourcing: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3880462/)
The intracellular signaling pathway of TLR4, including MyD88-dependent and MyD88-independent pathways, which can amplify stimulation signals and up-regulate inflammatory gene expression by recruiting downstream adaptor proteins.
The MyD88-dependent way is responsible for proinflammatory cytokine expression. Upon LPS stimulation, the intracellular TIR region of TLR4 binds to the carboxyl terminus of MAL and MyD88, while the amino terminus of MyD88 recruits IRAK4 (IL-1 receptor-associated kinase-4), IRAK1, IRAK2 through homotypic interactions [9]. The activated IRAK4, IRAK1, and IRAK2 are phosphorylated and detached from the MyD88/IRAK complex and bind to TRAF6 (TNF receptor-associated factor 6) [17]. TRAF6 is, in turn, activates TAK1 (Transforming growth factor B-activated kinase) complex. The activated TAK1 incites the IKKα/IKBKB complex, which phosphorylates the NFκB inhibitor (IκB) and then ubiquitination and degradation [18]. Free NFκB enters the nucleus to regulate transcription and activate gene expression of cytokines such as IL1, IL6, IL8, IL12[8], TNF-a, and IFN-y [10] [22]. At the same time, TAK1 can also activate the MAPK family complex, which activates AP1 (activator protein 1) and enters the nucleus to initiate gene expression.
It was shown that MyD88-deficient mice did not completely lose their response to LPS stimulation, and the activation of IRF3 and the expression of IFN-B were not affected, indicating that TLR4 signaling is not completely dependent on MyD88 Pathway.
In the MyD88-independent pathway, TLR4 recruits a TIR domain-containing TRAM (TRIF-related adaptor molecule) and TRIF, and TRIF activates the transcription factor IRF3 (interferon regulatory factor 3) [19] [20]. Activation of NF-κB and MAPK leads to delayed phase, ultimately inducing transcription of IFN1 (type I interferon) [11] and interferon-related genes, IFN-inducible protein 10, GARG-16 (glucocorticoid attenuated response gene 16), INF-regulated gene 1, IRG1 expression and dendritic cell maturation, promoting antiviral and antibacterial immunity [21]. The MyD88-independent pathway is also known as the TRIF-dependent pathway.
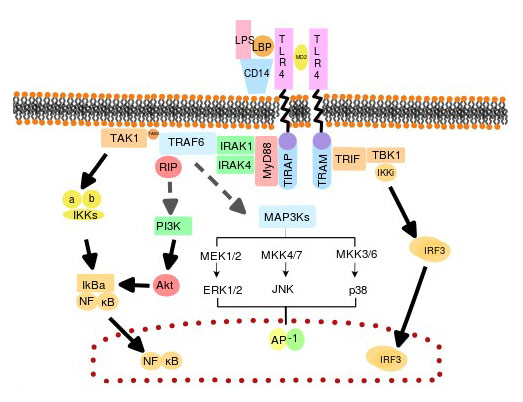
Figure 2 The MyD88-dependent pathway and MyD88-independent pathway
Osteoarthritis (OA) is characterized by articular cartilage and bone degeneration around the joints and secondary bone hyperplasia, which seriously affects the quality of life of patients. The study found that activation of the TLR4 signal transduction pathway is closely related to the production of catabolic products involved in joint destruction, which may participate in mediating the imbalance of OA cartilage metabolism. Bobaz K. Sunk IG, Hofstaeter G, etc. found TLR4 in the articular cartilage messenger mRNA. And its activation can lead to changes in the content of IL-1B, type II collagen and the like. In vitro studies have shown that TLR4 has an inhibitory effect on cartilage repair; TLR4 expression is up-regulated in cartilage injury sites in patients with advanced OA; Obawa T. Koyame K, etc. block TLR4 signal transduction pathway, and the symptoms of OA in mice are alleviated [23] [24] [25] [26].
Chronic inflammation is closely related to malignant tumors. The wide expression of TLR4 in tumors suggests that it plays an important role in the mechanism of chronic inflammation [28].
TLR4 signaling can effectively activate NFκB, and activation of NFκB is an important factor in the onset of chronic inflammation, which directly promotes the development of various tumors. Clinical pathological results uncover that TLR4 is not only highly expressed in Helicobacter pylori (HP)-positive gastric cancer tissues, but also in HP-positive gastric epithelial metaplasia and morphological abnormal tissues. And TLR4 signaling can significantly inhibit the proliferation of gastric cancer cell lines. TLR4 also up-regulates the expression of the inflammatory factors COX-2 and PGE2 (prostaglandin E2) by activating NFκB, which ultimately results in carcinogenesis of normal gastric epithelial cells. Similarly, human and mouse chronic colitis-associated colon tumor tissues also highly express TLR4. And experiments have shown that the incidence of colon cancer in TLR4 knockout mice is significantly lower than that in normal mice.
KELL.Y M G, ALVERO A B, CHEN R, et al. confirmed that the TLR4-MyD88-NFκB pathway promotes the proliferation of epithelial ovarian cancer cells. Other studies have shown that TLR4 activation can also induce the expression of some anti-apoptotic proteins, such as XIAP (a major inhibitor of caspase-3 and caspase-9) and phosphorylation of Akt, to promote the survival of ovarian cancer cells.
In addition, LPS promotes the proliferation of human prostate cancer cell line PC3 by activating NFκB and inducing expression of some cytokines (such as TGFB1, VEGF), and interfering with TLR4 expression inhibits proliferation of PC3 cells and induces apoptosis [29]. Induction of NFKB activation by the TLR4 signaling pathway also promotes inhibition of TNF-α and TRAIL-induced apoptosis in human lung cancer cell lines. The lung cancer specimens and cell line A549 studies showed that the degree of tumor cell differentiation was positively correlated with the expression level of TLR4, indicating that it may be involved in the development of lung cancer [30].
In seizures, TLR4 is abnormally expressed in nerve cells, astrocytes, and microglia. TLR4 acts as an important inflammatory receptor mediating epileptic inflammatory injury and is involved in the development and progression of epilepsy. In other words, seizures activate TLR4 and inflammatory factors. Activated TLR4 and inflammatory factors are involved in the production and development of epilepsy. Inflammatory factors can be mediated through the TLR4 signaling pathway, forming a cycle between the three paths. Furthermore, studies have shown that TLR4-deficient C3H/HeJ mice have a function of resisting seizures [31].
Cerebral cavernous malformations (CCMs) refer to spongy abnormal blood vessel masses composed of numerous thin-walled blood vessels. The actual disease is not a true tumor, but a vascular malformation that lacks arterial components. The cavernous vascular malformation is the cause of stroke and seizures, and there is no medical therapy. A 2016 study confirmed that the occurrence of familial and sporadic cavernous hemangioma is most likely due to the production of the MEKK3-KLF2/4 signaling pathway in endothelial cells.
In 2017, Alan T. Tang and others used LPS to activate TLR4 on the surface of cerebral vascular endothelial cells, which was found to greatly accelerate the formation of abnormal vascular clusters in the brain, further demonstrating that MEKK3-KLF2/4 signaling pathway is associated with cavernous hemangioma.
In the experiment, the researchers used genetic engineering techniques or drugs to block TLR4 signaling, effectively inhibiting the formation of cavernous vascular malformations in the mouse brain. Conversely, increased expression of TLR4 or its co-receptor CD14 gene promotes the formation of intracranial cavernous vascular malformations in mice. This study identified TLR4 as an upstream molecule that leads to MEKK3-KLF2/4 signaling activation.
On May 7, 2019, a study online published in journal Cell Reports, described that Cincinnati Children's scientists have discovered that one of the major risk factors of preterm birth originates from TLR4 in the endothelial cells in the lining of blood vessels of the decidua. And the findings may provide a potential therapeutic target for preventing preterm birth.
To mimic how infections trigger inflammation and further lead to preterm birth, the researchers used LPS to stimulate pregnant mice. They found that the mice response to LPS begins with the activation of TLR4 present on decidual vascular endothelial cells.
Activated TLR4 by LPS then promotes the release of a series of proinflammatory cytokines, including IL6. This process helps the body fight against infections. As IL6 elevates, IL10 releases to recover the inflammatory level. It keeps a balanced immunity. However, once TLR4 is excessively activated, IL10 cannot be enough to respond to restore the inflammatory level, which breaks the balanced immunity, causing a dangerous inflammation and a higher risk of preterm birth.
The team knocked out TLR4 in mice' decidual tissue and found that mice are highly sensitive to LPS, even at low doses, and are likely to experience preterm birth. And managing IL10 helps these mice avoid premature birth. However, deletion of TLR4 in vascular endothelial cells renders mice highly resistant to LPS-induced preterm birth, even at high doses.
These results indicate that systemic inflammation begins with TLR4 in decidual vascular endothelial cells, which is one of the high risks leading to preterm birth.
In conclusion, TLR4 is a peculiar member of the TLR family. And it is the only known TLR which uses all those adaptor proteins. TLR4 can specifically recognize and bind to LPS, eventually triggering a proinflammatory cascade. And the LPS/TLR4 signal transduction pathway is involved in many diseases, which potentially provides strategies to study and develop therapeutic ways or drugs for these related diseases.
References
[1] Hansson GK, Edfeldt K, et al. Toll to be paid at the gateway to the vessel wall [J]. Arterioscler Thromb. Vasc. Biol. 2005, 25 (6): 1085–7.
[2] Hashimoto C, Hudson KL, et al. The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein [J]. Cell. 1988, 52 (2): 269–79.
[3] Medzhitov R, Preston-Hurlburt P, et al. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity [J]. Nature. 1997, 388 (6640): 394–7.
[4] Fitzgerald, K. et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF [J]. J Exp Med. 2003, 198, 1043-1055.
[5] Medzhitov, R., Preston-Hurlburt, et al. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity [J]. Nature 388. 1997, 394-397.
[6] Hoshino, K, Takeuchi, O, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the LPS gene product [J]. Journal of Immunology. 1999, 162 (7): 3749–52.
[7] Poltorak, Alexander, et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene [J]. Science. 1998, 282 (5396): 2085–2088. .
[8]?Vaure C, Liu Y,?A comparative review of toll-like receptor 4 expression and functionality in different animal species [J].?Frontiers in Immunology. 2014,?5: 316.?
[9] Lu YC, Yeh WC, et al. LPS/TLR4 signal transduction pathway.?Cytokine. 2008,?42?(2): 145–51.
[10] P?lsson-McDermott EM, O'Neill LA.?Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4 [J].?Immunology. 2004,?113?(2): 153–62.
[11] O'Neill LA, Golenbock D, et al. The history of Toll-like receptors-redefining innate immunity [J].?Nature Reviews. Immunology. 2013,?13?(6): 453–60.?/p>
[12] Kawai, T. Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors [J].?Nat Immunol.2010,?11(5): 373–384.
[13] Triantafilou, M., K, et al. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster [J].?Trends Immunol. 2002, 23(6):301-4.
[14] da Silva Correia, J., K. Soldau, et al. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex: transfer from CD14 to TLR4 and MD-2 [J].?J Biol Chem. 2001,?276(24):21129-35.
[15] Miyake, K., Innate recognition of lipopolysaccharide by CD14 and Toll-like receptor 4-MD-2: unique roles for MD-2 [J].?Int Immunopharmacol. 2003,?3(1):119-28.
[16] Slack, J. L., K. Schooley, et al. Identification of two major sites in the type I interleukin-1 receptor cytoplasmic region responsible for coupling to pro-inflammatory signaling pathways [J].?J Biol Chem. 2000,?275(7):4670-8.
[17] Jiang, Z., M. Zamanian-Daryoush, et al. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFκB and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR [J].?J Biol Chem. 2003,?278(19):16713-9.
[18] Ghosh, S., M. Karin. Missing pieces in the NF-κB puzzle.?Cell. 2002,?109 Suppl:S81-96.
[19] Fitzgerald, KA, Palsson-McDermott EM, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction [J].?Nature 2001,?413(6851):78-83.
[20] Toshchakov V, Jones BW, et al TLR4, but not TLR2, mediates IFN-β-induced STAT1α/β-dependent gene expression in macrophages [J].?Nat Immunol.?3(8):392-8.
[21] Yamamoto, M., S. Sato, et al. Cutting Edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-β promoter in the Toll-like receptor signaling [J].?J Immunol. 2002,?169(12):6668-72.
[22] Salkowski, C. A., K. E. Thomas, et al.?Impaired IFN-γ production in IFN regulatory factor-1 knockout mice during endotoxemia is secondary to a loss of both IL-12 and IL-12 receptor expression [J].?J Immunol. 2000,?165(7):3970-7.
[23] Scanzello, CR,?Plaas A, et al.?Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound [J]? Curr Opin Rheumatol. 2008,?20(5):565–572.
[24] Steven R Goldring?&?Carla R Scanzello.?Plasma proteins take their toll on the joint in osteoarthritis [J].?Arthritis Research & Therapy, 2012,?14:111.
[25] Kikuchi T, Matsuguchi T,?et al.?Gene expression of osteoclast differentiation factor is induced by lipopolysaccharide in mouse osteoblasts via Toll-like receptors [J].?J. Immunol. 2001, 166(5):3574-9.
[26] Sohn, D. H.?et al.?Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4 [J].?Arthritis Research therapy, 2012, Ther.?14(1):R7.
[27] Lien, E, Means TK,?et al.?Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide [J].?J Clin Invest. 2000,?105(4):497–504.
[28] Hakim F, Wang Y, et al Fragmented sleep accelerates tumor growth and progression through recruitment of tumor-associated macrophages and TLR4 signaling [J].?Cancer Res.?2014, 749(5):1329–37.
[29] Lissbrant IF, Stattin P, et al. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival [J].?Int J Oncol.?2000, 17(3):445–51.
[30] Koukourakis MI, Giatromanolaki A, et al. Different patterns of stromal and cancer cell thymidine phosphorylase reactivity in non-small-cell lung cancer: impact on tumour neoangiogenesis and survival [J].?Br J Cancer.?1998, 77(10):1696–703.
[31] Farina C, Aloisi F, et al.?Astrocytes are active players in cerebral innate immunity [J].?Trends Immunol.?2007,?28(3):138-45.


Comments
Leave a Comment