Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
| Code | CSB-EP005400HU1 |
| Abbreviation | Recombinant Human CHRNE protein, partial |
| MSDS | |
| Size | $306 |
| Order now | |
| Image |
|
| Have Questions? | Leave a Message or Start an on-line Chat |
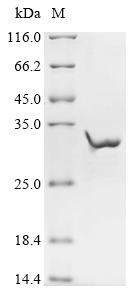
The expression region of this recombinant Human CHRNE covers amino acids 21-239. This CHRNE protein is theoretically predicted to have a molecular weight of 32.5 kDa. This protein is generated in a e.coli-based system. The CHRNE coding gene included the N-terminal 10xHis tag and C-terminal Myc tag, which simplifies the detection and purification processes of the recombinant CHRNE protein in following stages of expression and purification.
The human acetylcholine receptor subunit epsilon (CHRNE) is a key component of the nicotinic acetylcholine receptor (nAChR), which is a ligand-gated ion channel found at neuromuscular junctions. CHRNE is predominantly expressed in skeletal muscle and plays a crucial role in neurotransmission by responding to acetylcholine released from nerve terminals. Upon acetylcholine binding, the nAChR undergoes conformational changes, leading to the influx of cations and muscle cell depolarization, ultimately initiating muscle contraction. Mutations in CHRNE can lead to congenital myasthenic syndromes (CMS), a group of genetic neuromuscular disorders characterized by muscle weakness and fatigue. Studying CHRNE helps unravel the mechanisms of neuromuscular communication and aids in understanding and treating related disorders.There are currently no reviews for this product.

