Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Parkinson's disease (PD) was first described in 1817 by the British Dr. James Parkinson in his "An Essay on the Shaking Palsy", where he described six patients suffered from a neurological disorder characterized by emaciation and quivering [1]. Seventy years later, the French neurologist Jean-Martin Charcot presented the first clinical description of the disorder, bestowing the eponymous name "Parkinson's disease" as a tribute to James Parkinson.
Parkinson's disease (PD) is a chronic, progressive neurological disorder and has become the second most common neurodegenerative disease worldwide, affecting 1% of the population over the age of 65 [2].
Parkinson's disease is primarily divided into sporadic and familiar forms. The majority (90-95%) of patients have sporadic PD, also known as idiopathic PD (IPD), which is determined by the complicated interplay between the individual's genetic constitution and environmental factors. Familial cases, resulting from Mendelian inheritance of genetic mutations, account for around 5-10% of Parkinson's disease patients. Individuals carrying PD-related genes have a higher risk for PD.
The onset of Parkinson's disease symptoms is typically gradual and mild initially. There is a variety of symptoms associated with the disease, but their progression and severity vary among individuals. It's uncommon for a person with Parkinson's to encounter all or most of these symptoms.
Core motor symptoms of Parkinson's disease include rigor and tremor, rigidity, bradykinesia, postural instability, and walking or gait difficulty.
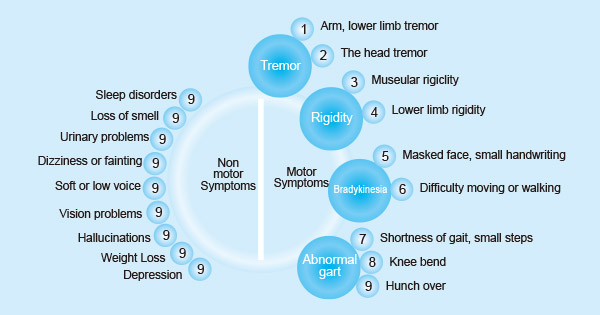
Figure 1. Symptoms of Parkinson's disease
Apart from motor symptoms, individuals with PD commonly contend with associated non-motor symptoms. This underscores that PD is not exclusively a disorder of the central nervous system but rather a systemic condition.
The causes of Parkinson's disease are not fully understood, but substantial studies have shown that the pathology of Parkinson's disease is the degeneration of substantia nigra (SN) dopaminergic neurons and the presence of Lewy bodies containing α-synuclein (α-syn) aggregates [5-7]. The aggregation and accumulation of α-syn perpetuate neuronal degeneration.
Research also suggests the etiology of Parkinson's disease is linked to multiple factors, including genetic susceptibility, environmental factors, aging, and so on.
At least 5% of PD cases are associated with mutations in many genes, including leucine-rich repeat kinase 2 (LRRK2), α-synuclein (SNCA), glucosylceramidase β 1 (GBA1), phosphatase and tensing homolog-induced kinase 1 (PINK1), parkin (PRKN), and PARK7 (DJ-1) [8-10]. These genes have been linked to an elevated risk of familial PD.
These mutated gene products result in abnormalities in intracellular trafficking, oxidative stress, mitochondrial dysfunction, disrupted protein quality control, protein misfolding and aggregation, dysfunction of the ubiquitin-proteasome system, and modified kinase activity, playing a key role in the pathogenesis of PD. Certain mutations may also contribute to the early onset of PD.
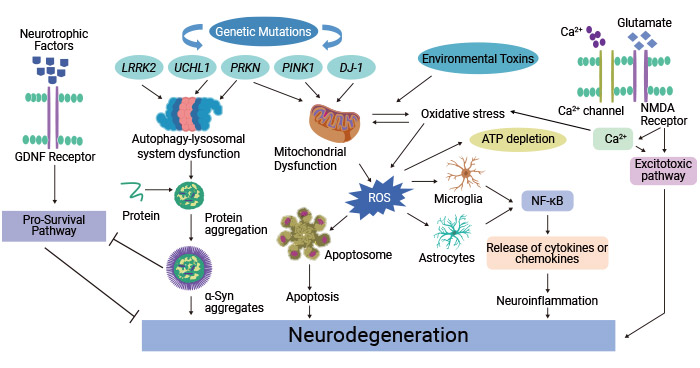
Figure 2. Molecular mechanism involved in the pathophysiology of Parkinson's disease
The picture is cited from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8962417/
| PARK Locus | Gene/Protein | Inheritance | Physiological Function |
|---|---|---|---|
| PARK1/4 | SNCA/α‐synuclein | Autosomal dominant (AD) | Clathrin‐mediated endocytosis Neurotransmitter release Exosome release Chaperone‐mediated autophagy The main component of Lewy bodies (LB), and α-syn aggregation and accumulation perpetuate neuronal degeneration |
| PARK5 (putative) | UCHL1 | AD | Ubiquitin C-Terminal Hydrolase L1: processing ubiquitinated proteins and ubiquitin precursors |
| PARK8 | DARDARIN/LRRK2 | AD | Clathrin‐mediated endocytosis Autophagy Neurotransmitter release Endo‐lysosomal trafficking Exosome release Monogenic pathogenic mutations either reduce GTPase activity or increases kinase activity |
| PARK11(putative) | GIGYF2 | AD | GRB10-interacting GYF protein 2: repressing translation initiation |
| PARK13(putative) | HTRA2 | HtrA Serine Peptidase 2: proteolytic activity and promotes apoptosis | |
| PARK17 | VPS35 | AD | Autophagy Endo‐lysosomal trafficking Golgi complex trafficking VPS35 mutation leads to retromer dysfunction, protein aggregation, mitochondrial dysfunction, disturbed dopamine signaling, blocked plasma membrane receptor recycling and vesicular trafficking [11] |
| PARK18 | EIF4G1 | AD | Controls the commencement of the translation of mRNAs encoding mitochondrial, cell survival, and growth proteins |
| PARK21 | DNAJC13/RME‐8 | AD | Clathrin‐mediated endocytosis Endosomal sorting/trafficking Pathogenic DNAJC13 mutation induces abnormal endosomal retention of α-synuclein |
| PARK22 | CHCHD2 | AD | Involved in the mitochondrial respiratory chain complex; the T61I mutation in CHCHD2 causes an autosomal dominant form of PD |
| PARK2 | PRKN/Parkin | Autosomal recessive (AR) | Clathrin‐mediated endocytosis Mitochondrial quality control neurotransmitter release Ubiquitination, regulate the autophagic degradation of mitochondria Tumorigenesis |
| PARK6 | PINK1 | AR | Mitochondrial quality control (mitophagy, fusion and fission, mitochondrial derived vesicles) |
| PARK7 | PARK7/DJ‐1 | AR | Mitochondrial function mitochondrial quality control of reactive oxygen species transcription regulation |
| PARK9 | ATP13A2 | AR | Endo‐lysosomal pathway Exosome release Associated with juvenile-onset PD Kufor-Rakeb syndrome (KRS); mutation of ATP13A2 leads to lysosomal dysfunction, causing abnormal buildup of α-synuclein [12] |
| PARK14 | PLA2G6 | AR | Membrane trafficking Phospholipid metabolism Mitochondrial function |
| PARK15 | FBX07 | AR | Ubiquitination and proteasomal degradation Proliferation |
| PARK19 | DNAJC6/Auxilin | AR | Clathrin‐mediated endocytosis Golgi–lysosome trafficking |
| PARK20 | SYNJ1/Synaptojanin 1 | AR | Clathrin‐mediated endocytosis Synaptic autophagy |
| PARK23 | VPS13C | AR | Lipid transport protein implicated in mitochondrial biogenesis and mitophagy |
The Table information is sourced from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6138432/ and https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10308076/
Nervous system problems: Supranuclear palsy, Wilson's disease, Huntington's disease, Haller Walden-Spartz syndrome, and Alzheimer's disease can also cause Parkinson's disease.
Brain injury: Traumatic brain injury, which causes changes in consciousness levels, increases the risk of PD in the years following injury [13].
Living area: There are differences in the geographical distribution of PD. This may be due to differences in environmental and genetic risk factors.
Occupations: Certain occupational categories or titles are associated with higher rates of PD. Stress at work increases the risk of Parkinson's disease.
Pesticide exposure: Pesticides (including cypermethrin and hexachlorocyclohexane) have been reported most consistently in all chemical exposures associated with Parkinson's disease.
Contact with metals: Occupational exposure to various metals is considered to be related to the development of PD.
Age is the biggest risk factor for developing Parkinson's disease, with the incidence increasing significantly at the age of 60 and exponentially in the subsequent decades of life. Parkinson's disease mainly affects the elderly, especially those between the ages of 55 and 65, with 1% of people over 65 years old and 5% or more of people over 85 years old affected [14].
Young Onset Parkinson's Disease (YOPD) occurs in people under 50 years of age. In rare cases, Parkinson-like symptoms may occur in children and adolescents. People diagnosed with YOPD have a more frequent family history of Parkinson's disease and a longer survival period.
Epidemiology showed a prevalence rate of 15 ~ 328/100,000 and about 1% of > 65-year-old population. The incidence is 10 to 21 per 100,000 population per year. In the United States, the incidence is 21 cases per 100,000 people [15]. Given the increase in global life expectancy, the number of people affected by Parkinson's disease and the consequent personal, social, and economic burden are expected to increase dramatically by 2030 [16]. By 2020, nearly one million people in the United States will have Parkinson's disease, and about 60,000 Americans are diagnosed with Parkinson's disease each year. More than 10 million people worldwide suffer from Parkinson's disease.
Age: Parkinson's disease occurs mainly in the elderly, although Parkinson's can be developed among young people, it is estimated that only 4% of people with Parkinson's disease were diagnosed before the age of 50. The average age of onset of Parkinson's disease is 60 years, and the prevalence of PD increases with age.
Gender: Men are 1.5 times more likely than women to develop Parkinson's disease. People with Parkinson's disease in their parents or siblings may double their chance of developing Parkinson's disease because of genetic mutations.
Race: There is a racial difference in prevalence, with whites being the highest, followed by yellows and blacks being the lowest.
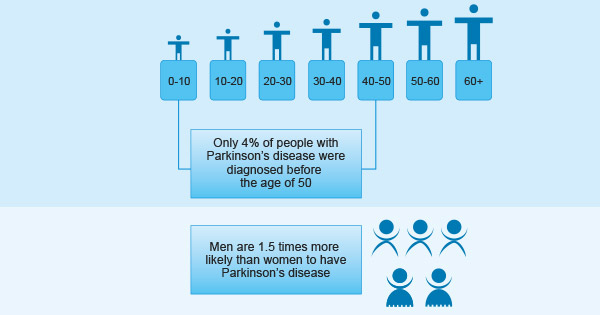
Figure 3. Facts about Parkinson's disease
When famous people disclose their illness, it helps raise people's attention to the disease and increase their awareness and understanding of the disease.
Muhammad Ali (diagnosed in 1984) was diagnosed with Parkinson's disease three years after he left the boxing campaign. He has raised awareness of Parkinson's disease worldwide and helped establish the Muhammad Ali Parkinson Center in Phoenix, Arizona.
Michael j fox (diagnosed in 1991) is one of the world's most famous patients with Parkinson's disease. He devoted his life to the further development of Parkinson's research and established the Michael j fox Parkinson Research Foundation.
Brian Grant (diagnosed with Parkinson's disease in 2008) and established the Brian Grant Foundation to help others do the same.
In addition, George H. Bush, Billy Connolly, Alan Arda, Neil Diamond, and Pat Torpey were also diagnosed with Parkinson's disease.
Although the exact cause of Parkinson's disease is not fully understood, several molecular pathways and processes are believed to play a role in the development and progression of this disease. Understanding these pathways is crucial for developing targeted therapies aimed at slowing or halting the progression of Parkinson's disease.
Pathologically, PD is mainly featured in the depletion of dopaminergic neurons in the substantia nigra pars compacta (SNPC) projecting to the striatum along the nigrostriatal pathway. The nigrostriatal pathway is an important dopaminergic pathway responsible for regulating voluntary movements and motor function.
This neurodegeneration leads to a significant loss of dopamine levels in the striatum, which increases the overall excitatory drive in the basal ganglia, blocking voluntary motor control and causing PD-specific motor impairments [17]. Patients typically manifest the PD motor symptoms when dopaminergic neurons lose up to 50% to 80%.
Alpha-synuclein, a presynaptic protein comprising 140 amino acids, exhibits multiple conformations and exists in various oligomeric states, maintaining a dynamic equilibrium. Mutant α-synuclein undergoes conformational changes, making it susceptible to the formation of aggregates and Lewy bodies. The aggregation of αlpha-synuclein is believed to be neurotoxic and contributes to the progressive degeneration of dopaminergic neurons in the substantia nigra. This neurodegeneration leads to the characteristic motor symptoms of Parkinson's disease.
Mutations in genes SNCA, LRRK2, VPS35, CHCHD2, PARK, PINK1, DJ-1, ATP13A2, PLA2G6, and FBXO7, are related to mitochondrial dysfunction in PD. PINK1 and Parkin mutation are associated with mitophagy and can cause the buildup of defective mitochondria and contribute to dysfunction.
Mitochondrial dysfunction in PD leads to energy production deficits (especially in dopaminergic neurons), increased oxidative stress, damage to mitochondrial DNA, apoptosis, altered calcium homeostasis, impaired mitophagy, α-synuclein accumulation, and overall cellular dysfunction. All these ultimately accelerate the degeneration and death of dopaminergic neurons.
Oxidative stress is a key player in the pathogenesis of PD. In PD, this oxidative damage is particularly detrimental to dopaminergic neurons in the substantia nigra. Mitochondrial dysfunction, α-synuclein aggregation, and inflammation contribute to increased reactive oxygen species (ROS) production in PD. Oxidative stress can further exacerbate mitochondrial impairment, trigger apoptotic pathways, and promote α-synuclein misfolding.
Neuroinflammation serves as a protective mechanism for restoring normal brain structure and function. However, when excessively activated, neuroinflammation becomes a significant driver of neurodegeneration. Chronic neuroinflammation stands out as a crucial hallmark in the pathology of PD.
Neuroinflammation is characterized by the activation of immune cells like microglia and astrocytes in the brain. The immune response is triggered by factors such as α-synuclein aggregation, mitochondrial dysfunction, and oxidative stress. Activated microglia release inflammatory mediators, contributing to a chronic inflammatory environment in affected areas, especially the substantia nigra. This sustained immune response can exacerbate neuronal damage and contribute to the progression of PD.
Autophagy plays a crucial role in the clearance of misfolded proteins, particularly α-synuclein, whose aggregation is a hallmark of PD pathology. Neurons, particularly dopaminergic neurons affected in PD, heavily rely on efficient autophagy for maintaining cellular homeostasis. Autophagy dysfunction can compromise neuronal health and contribute to the progressive degeneration observed in PD.
Mutations in genes related to autophagy, such as PINK1 and Parkin, have been associated with familial forms of PD. These genes are crucial for maintaining proper autophagic function. Autophagy relies on lysosomes for the degradation of engulfed cellular material. Dysfunction in lysosomal pathways can impair the autophagic process and contribute to the accumulation of toxic protein aggregates in PD.
In summary, PD is a progressive neurodegenerative disorder pathologically marked by dopaminergic neuron demise in the substantia nigra pars compacta and the formation of α-synuclein-containing Lewy bodies. Multiple factors work together to contribute to the development of PD, including α-synuclein aggregation that forms Lewy bodies, mitochondrial dysfunction, neuroinflammation, as well as excitotoxicity and metal accumulation.
Current research on Parkinson's disease emphasizes the interplay between genetic and environmental factors, the propagation of PD pathology from the periphery to the central nervous system, and a detailed exploration of the molecular mechanisms underlying neuroinflammation and the degeneration of dopamine neurons.
References
[1] Parkinson T. Outlines of Zoonosological Tables. Lond. Med. Phys. J. 1817;38:449–453.
[2] Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson's Disease [J]. J. Neural Transm. 2017, 124, 901–905.
[3] Siciliano Mattia, Trojano Luigi, Santangelo Gabriella. et al. Fatigue in Parkinson's disease: A systematic review and meta-analysis [J]. Mov. Disord, 2018, undefined: undefined.
[4] Swann Peter, O'Brien John T. Management of visual hallucinations in dementia and Parkinson's disease [J]. Int Psychogeriatr, 2018, undefined: 1-22.
[5] Trist B., Hare D., Double K. (2019). Oxidative stress in the aging substantia nigra and the etiology of Parkinson's disease [J]. Aging cell 18:e13031.
[6] Alvarez-Erviti L., Seow Y., et al. (2011). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission [J]. Neurobiol. Dis. 42 360–367. 10.1016/j.nbd.2011.01.029.
[7] Roberts R., Wade-Martins R., Alegre-Abarrategui J. (2015). Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinson's disease brain [J]. Brain 138 1642–1657.
[8] Kim J., Daadi E.W., Oh T., Daadi E.S., Daadi M.M. Human Induced Pluripotent Stem Cell Phenotyping and Preclinical Modeling of Familial Parkinson's Disease [J]. Genes. 2022;13:1937.
[9] Verstraeten A., Theuns J., Van Broeckhoven C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era [J]. Trends Genet. 2015;31:140–149.
[10] Kalinderi K., Bostantjopoulou S., Fidani L. The genetic background of Parkinson's disease: Current progress and future prospects [J]. Acta Neurol. Scand. 2016;134:314–326.
[11] Rahman AA, Morrison BE. Contributions of VPS35 Mutations to Parkinson's Disease [J]. Neuroscience. 2019 Mar 1;401:1-10.
[12] Zhang F, Wu Z, Long F, et al. The Roles of ATP13A2 Gene Mutations Leading to Abnormal Aggregation of α-Synuclein in Parkinson's Disease. Front Cell Neurosci. 2022 Jul 6;16:927682.
[13] Gardner Raquel C, Byers Amy L, Barnes Deborah E, et al. Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study [J] .Neurology, 2018, 90: e1771-e1779.
[14] Pringsheim T, Jette N, Frolkis A, et al. The prevalence of Parkinson's disease: a systematic review and meta-analysis [J]. Movement Disorders Official Journal of the Movement Disorder Society, 2015, 29(13):1583-1590.
[15] Savica R, Grossardt BR, Bower JH, et al. Incidence and pathology of synucleinopathies and tauopathies related to parkinsonism [J]. JAMA Neurol, 2013, 70:859-66.
[16] Dorsey ER, Constantinescu R, Thompson JP, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030 [J]. Neurology, 2007; 68:384-6.
[17] Gordián-Vélez WJ, Chouhan D, et al. Restoring lost nigrostriatal fibers in Parkinson's disease based on clinically-inspired design criteria [J]. Brain Res Bull. 2021 Oct;175:168-185.


Comments
Leave a Comment