Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
When acclaimed actress Kathy Bates received her thyroid cancer diagnosis, she joined the millions navigating a disease where uncertainty often reigns. Her successful treatment highlights a critical truth: thyroid tumor management is undergoing a revolution. Gone are the days of one-size-fits-all approaches. Today, the frontier lies in deciphering the unique molecular blueprint of each tumor, enabling a shift from invasive procedures to precise, personalized strategies that maximize efficacy and minimize impact.
Thyroid tumors are a biologically diverse group of lesions that range from indolent nodules to aggressive cancers. For researchers, the main challenge is not only classification, but also understanding how genetic changes, tumor evolution, and metabolic shifts shape disease behavior. This article summarizes the core biology of thyroid tumors, focusing on mechanisms of progression, metabolic features, and emerging drug targets [1-4].
Table of Contents
1. Background on Thyroid Tumors
2. Major Categories of Thyroid Tumors
3. Mechanisms of Thyroid Tumor Progression
Thyroid tumors are common in endocrine practice, yet their biology is highly variable. Some lesions remain stable for years, while others progress through distinct molecular and pathologic states. A clear research framework begins with the basic tumor origin, pathology, and the reasons these lesions matter for translational science.
Thyroid tumors arise mainly from follicular cells (thyroid hormone synthesis) or, less commonly, parafollicular C cells (calcitonin production). Follicular cell-derived tumors include benign adenomas and differentiated thyroid cancers such as papillary and follicular carcinoma, while C-cell tumors include medullary thyroid carcinoma. This distinction is important because the cell of origin strongly influences both biology and treatment strategy [1,4].
Thyroid tumors are useful models for studying cancer initiation, differentiation, and progression. They also provide a strong example of how a tumor can shift from a well-differentiated state to a more aggressive phenotype through the accumulation of molecular changes. For researchers, this makes thyroid cancer a practical system for studying tumor heterogeneity and precision oncology [1,3].
The latest World Health Organization (WHO) 5th edition classifies thyroid tumors mainly by cell of origin and by clinical risk (benign, low‑risk, malignant) [1,5]. Follicular cell‑derived neoplasms are grouped separately from C‑cell–derived tumors and other rare entities, reflecting both biology and prognosis.
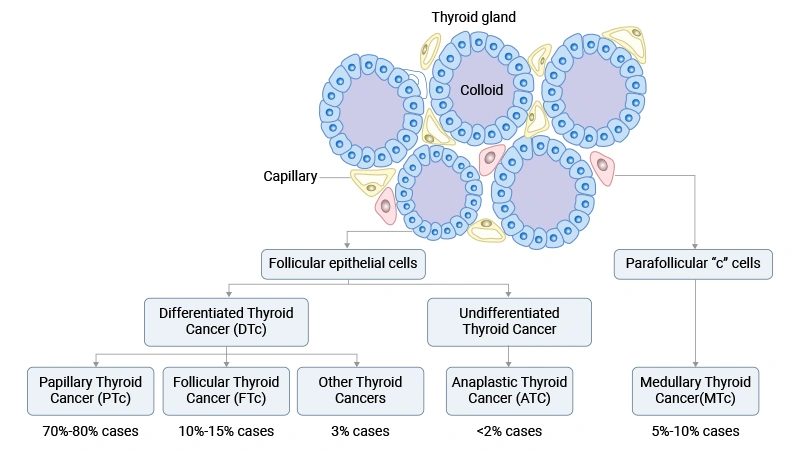
Figure. Types of thyroid cancer
Follicular cell‑derived neoplasms are the most common thyroid tumors and are now explicitly divided into benign, low‑risk, and malignant categories [1,5,6].
Benign tumors arise from follicular epithelial cells but do not show invasive or metastatic behavior.
- Includes hyperplastic nodules in multinodular goiter.
- Often polyclonal and reactive rather than true neoplasms [1,5].
- Encapsulated, non‑invasive follicular neoplasm.
- Lacks capsular or vascular invasion and does not metastasize [1].
- Benign tumor composed of oncocytic (Hürthle) cells with granular eosinophilic cytoplasm.
- Shows mitochondria‑rich cytoplasm but no invasion [1].
These lesions are clinically important because they can mimic low‑risk or malignant tumors morphologically.
Low‑risk neoplasms are biologically neoplastic but have very limited malignant potential, with excellent outcomes when correctly managed [1,5].
- Encapsulated or well‑demarcated follicular tumor with papillary‑type nuclear changes but no invasion.
- Reclassified from carcinoma to reduce overtreatment [1,7].
- Tumor with equivocal or minimal capsular/vascular invasion that is not sufficient to diagnose carcinoma.
- Used when pathologic criteria for clear malignancy are not fully met [1,5].
- Rare trabecular lesion with hyalinized stroma and PTC‑like nuclear features.
- Considered low‑risk despite its worrisome appearance [1,7].
These entities highlight the effort to separate biologically indolent tumors from clearly malignant disease.
Malignant follicular cell‑derived tumors show invasive behavior and varying metastatic potential. They are further stratified by differentiation and grade [1,5].
- The most common thyroid carcinoma accounts for 70% to 80% of thyroid cancers.
- Defined by characteristic nuclear features (enlargement, grooves, pseudoinclusions) and often associated with BRAF or RET pathway alterations [1].
- Includes multiple variants (classic, follicular variant, tall cell, etc.) in detailed WHO tables [1].
- Accounts for less than 15% of all thyroid cancers.
- Typically associated with RAS mutations or PAX8::PPARG fusions, and maintains follicular differentiation [1,8].
- Shows follicular architecture and capsular and/or vascular invasion.
- Malignant oncocytic (Hürthle) cell tumor with capsular or vascular invasion.
- Often has distinct mitochondrial and genetic features compared with non‑oncocytic FTC [1].
- Occupy an intermediate position between well‑differentiated carcinoma and anaplastic carcinoma [5].
- Diagnosed based on high mitotic activity and/or tumor necrosis; PDTC also meets Turin criteria for solid/trabecular/insular growth [5].
- Carry a worse prognosis than classic PTC/FTC but are not as aggressive as anaplastic carcinoma.
- Highly aggressive, undifferentiated carcinoma, usually arising in older adults.
- Represents a small fraction (less than 2%) of thyroid cancers but accounts for a disproportionate number of thyroid cancer deaths.
- Histology shows marked pleomorphism, high mitotic rate, and necrosis [7].
This graded system emphasizes that thyroid carcinoma spans a continuum from well‑differentiated, low‑risk tumors to undifferentiated, highly lethal disease.
Medullary Thyroid Carcinoma (MTC)
Medullary thyroid carcinoma arises from parafollicular C cells, not follicular cells, and is therefore classified separately [1,5].
Because of its distinct biology and genetics, MTC is managed and studied as a separate disease entity.
Mixed Medullary and Follicular Cell-Derived Thyroid Carcinomas
Mixed carcinomas contain both medullary (C‑cell) and follicular‑derived components [1].
In addition to follicular and C‑cell tumors, the WHO classification describes several less common groups.
These tumors resemble salivary gland neoplasms histologically and genetically.
Both entities are rare but important because they broaden the differential diagnosis for unusual thyroid masses [1,7].
Some thyroid tumors do not clearly fit into follicular or C‑cell lineages.
These entities may show unique molecular profiles (e.g., association with familial adenomatous polyposis in cribriform‑morular carcinoma) and are grouped as “tumors of uncertain histogenesis” in the WHO 5th [1,5].
Thymic epithelial tumors can occur within or adjacent to the thyroid and are classified separately.
These lesions reflect ectopic or remnant thymic tissue and are recognized as a distinct category in the updated WHO scheme [1,5].
The WHO 5th edition also lists embryonal thyroid neoplasms, including thyroblastoma, as a separate group.
Thyroid tumor progression is a multistep process driven by oncogenic pathway activation, clonal evolution, dedifferentiation, epigenetic remodeling, and microenvironmental selection. Aggressive thyroid cancer emerges when early driver events are joined by additional changes that increase plasticity, reduce differentiation, and promote invasion.
Most follicular-derived thyroid cancers begin with alterations in the MAPK pathway, especially BRAF, RAS, and RET/PTC events. These changes act as early oncogenic triggers and help explain why many papillary thyroid carcinomas share a common biological backbone. In more advanced tumors, additional abnormalities accumulate, including changes that affect the PI3K-AKT pathway and tumor suppressor genes [4,9,10].
The MAPK pathway is one of the central signaling routes in thyroid tumorigenesis. When BRAF V600E or RAS is activated, the pathway promotes proliferation, survival, and altered differentiation. This helps drive the transition from a normal thyroid follicular cell to a malignant clone [9,10].
The PI3K-AKT pathway is especially important in tumor progression and in tumors with follicular growth patterns. It supports cell survival, metabolic adaptation, and invasive behavior. In aggressive thyroid cancers, MAPK and PI3K-AKT signaling often work together rather than separately [4,10].
As thyroid tumors progress, they tend to accumulate more genetic alterations. This creates clonal evolution, where one dominant clone expands while minor subclones emerge under selection pressure. The result is a more heterogeneous tumor with greater potential for invasion, relapse, and therapy resistance [4,9,10].
One of the most important progression mechanisms is dedifferentiation, in which tumor cells lose normal thyroid features and become less specialized. This process is closely linked to more aggressive histology and reduced responsiveness to radioiodine treatment. It marks a shift from differentiated thyroid cancer toward poorly differentiated or anaplastic disease [4,10].
As tumors dedifferentiate, they often downregulate thyroid-specific genes involved in iodine uptake and hormone synthesis. This loss makes the tumor less responsive to standard thyroid-directed therapies. The biological effect is important because it changes both tumor behavior and treatment options [4].
Dedifferentiation is often accompanied by more mitotic activity, necrosis, invasion, and broader molecular disruption. In some cases, papillary or follicular tumors may progress into poorly differentiated thyroid carcinoma or anaplastic thyroid carcinoma. This transition represents one of the clearest examples of progression in thyroid oncology [4,9].
Progression is not driven only by DNA mutations. Epigenetic change, including altered DNA methylation and transcriptional reprogramming, also contributes to malignant evolution. These changes can silence differentiation genes and activate programs that support survival and invasion [4,9].
Aberrant DNA methylation can reduce the expression of tumor suppressors and thyroid differentiation genes. This helps lock tumor cells into a more aggressive state. In advanced disease, epigenetic silencing can reinforce the effects of oncogenic signaling [4,9].
Oncogenic signaling changes the transcriptional program of thyroid cells, shifting them toward proliferation, invasion, and stress resistance. This rewiring affects many downstream pathways, not just the original driver mutation. The tumor, therefore, behaves as a remodeled system rather than a simple mutation-bearing cell population [4,10].
The tumor microenvironment also shapes progression. Stromal cells, immune cells, and local tissue conditions can either restrain or promote tumor growth. In thyroid cancer, these signals help determine which clones survive and expand [4,10].
Fibroblasts, immune cells, and vascular elements can support invasion by creating a permissive tissue niche. These interactions may promote angiogenesis, immune evasion, and local spread. The microenvironment is therefore part of the progression machinery, not just a background feature [4].
Low oxygen and other stress conditions can favor more aggressive subclones. Under stress, cancer cells adapt by reprogramming metabolism and activating survival pathways. This helps explain why advanced thyroid tumors often become more robust over time [4].
The result of these interacting mechanisms is progression to poorly differentiated or anaplastic thyroid cancer. At this stage, tumors show greater invasiveness, resistance to standard therapy, and worse clinical outcomes. The biological shift reflects layered changes in signaling, genome stability, differentiation, and tissue context.
Thyroid cancer is not only a disease of mutations and cell growth. It is also a disease of altered metabolism. Thyroid cancer shows clear metabolic reprogramming, especially increased glycolysis, altered lipid synthesis, and changed amino acid use. These changes support tumor growth, help cells adapt to stress, and may create new diagnostic and therapeutic opportunities. For researchers, metabolism is now a central part of understanding how thyroid cancer develops and how it might be controlled.
Thyroid cancer cells often rewire their metabolism to support rapid proliferation and stress resistance. A central theme in the literature is the shift from normal oxidative metabolism toward aerobic glycolysis, a pattern often called the Warburg effect. In this state, tumor cells take up more glucose and convert more of it to lactate, even when oxygen is available [11-13].
One of the best-known metabolic changes in thyroid cancer is increased glucose uptake. Studies report upregulation of glucose transporters such as GLUT1, along with higher glycolytic enzyme activity. This helps explain why advanced or poorly differentiated thyroid tumors often show stronger uptake on FDG-PET imaging [12,13].
Thyroid cancer cells frequently depend more on glycolysis for energy and biosynthetic precursors than normal thyroid cells do. This metabolic pattern is not only about ATP production; it also supplies carbon skeletons for nucleic acids, proteins, and lipids. In practical terms, glycolysis helps the tumor build the materials it needs to expand [12-14].
Thyroid tumors also alter lipid synthesis and amino acid use. Recent studies show that metabolic remodeling in thyroid cancer extends beyond glucose metabolism and includes changes in fatty acid production, glutamine use, and one-carbon metabolism. These pathways are important because they support both growth and redox balance.
Cancer cells often increase de novo lipid synthesis, meaning they make lipids from scratch rather than depending only on uptake from the environment. In thyroid cancer, this can support membrane formation and signaling. Lipid metabolism may also vary by tumor subtype, which suggests that different thyroid cancers may have different metabolic dependencies [14-16].
Glutamine and other amino acids can feed the tricarboxylic acid cycle, support nucleotide synthesis, and help control oxidative stress. Multi-omics studies suggest that some thyroid cancer subtypes have distinct amino acid and urea cycle patterns, which may reflect different growth strategies. This is important for researchers looking for subtype-specific biomarkers or targets [2,14,15].
Thyroid cancer metabolism is shaped not only by tumor cells, but also by surrounding stromal cells. Reviews describe a form of metabolic coupling in which cancer-associated fibroblasts and tumor cells exchange fuels and metabolites. This interaction can help create a more supportive environment for tumor growth.
The reverse Warburg effect refers to a process in which stromal fibroblasts shift toward glycolysis and provide metabolic intermediates that tumor cells can use. In this model, the microenvironment does not simply respond to the tumor; it actively supports it. That makes metabolic research in thyroid cancer more complex than studying cancer cells alone [11,17].
Low oxygen and nutrient-poor conditions can further push thyroid cancer cells toward metabolic flexibility. Under these conditions, cells adapt by altering glycolysis, mitochondrial activity, and antioxidant systems. This helps explain why aggressive tumors can survive in hostile tissue environments [12-14].
Not all thyroid cancers share the same metabolic profile. Metabolomic and multi-omics studies indicate that papillary, follicular, medullary, and poorly differentiated tumors can show distinct metabolic fingerprints. These differences matter because they may guide more tailored research and treatment design.
Papillary thyroid carcinoma often shows strong glycolytic activity and pathway activation linked to oncogenic signaling, such as MAPK. In many cases, these tumors retain some differentiation, but their metabolic profile can still indicate aggressive behavior [11,12,14].
Follicular-patterned and more aggressive thyroid cancers may show broader metabolic remodeling, including glutamine use and altered lipid pathways. As tumors dedifferentiate, they often lose normal thyroid functions and gain more flexible energy metabolism [2,12,14,15].
In all, the metabolic features of thyroid cancer are not just descriptive. They may also be useful clinically. Imaging, biomarker development, and therapeutic targeting all depend on a better understanding of tumor metabolism. This is why metabolism is becoming a practical part of thyroid cancer research [11,12,17].
Several reviews suggest that enzymes, transporters, and metabolic pathways could become treatment targets in thyroid cancer. These include glucose transport, glycolysis, lipid synthesis, and glutamine-related pathways. While many of these strategies are still experimental, they point toward a more mechanism-based approach to therapy [12,14,17].
Therapy is becoming more precise as the molecular basis of thyroid cancer becomes clearer. Targeted drugs now focus on specific drivers such as RET and BRAF, while other strategies aim to restore differentiation or block resistance pathways.
RET is a major target in both medullary thyroid carcinoma and some papillary thyroid cancers with RET fusions. Selective RET inhibitors such as selpercatinib and pralsetinib have shown strong activity and better tolerability than older multikinase inhibitors. For researchers, this is a strong example of genotype-guided therapy in thyroid oncology [3,18].
In BRAF V600E-mutant anaplastic thyroid cancer, the combination of BRAF and MEK inhibition, such as dabrafenib plus trametinib, has produced meaningful responses. These treatments work by blocking a major growth pathway, but resistance can still develop over time. This makes BRAF-mutant disease a useful model for studying adaptive resistance [3,18].
Some thyroid cancers lose the ability to take up iodine, which reduces the effect of radioiodine therapy. MAPK inhibition can sometimes restore thyroid-specific gene expression and improve iodine handling, a strategy called redifferentiation. This approach is important because it aims not just to kill tumor cells, but to recover a lost function that makes standard therapy work better [3].
Metabolic enzymes and transporters are gaining attention as potential targets in advanced thyroid cancer. For example, studies have identified vulnerabilities in amino acid transport and one-carbon metabolism, including SLC7A5 and SHMT2-related pathways. These targets are still being developed, but they point toward a broader view of treatment that includes both signaling and metabolism [2,14].
Thyroid tumors are diverse, and their behavior is shaped by cell of origin, driver mutations, evolutionary pressure, and metabolic state. For researchers, the most useful view is a layered one: pathology defines the lesion, molecular biology explains progression, metabolism reveals dependencies, and targeted therapy turns that knowledge into action.
The Metabolic Phenotype of Tumor-Associated Macrophages in the Tumor Microenvironment
How the Tumor Microenvironment Fuels Cancer's Deadly Evolution
References
[1] Chiba, T. (2024). Molecular Pathology of Thyroid Tumors: Essential Points to Comprehend Regarding the Latest WHO Classification [J]. Biomedicines, 12(4), 712.
[2] Kim YH, Yoon SJ, et al. Integrative Multi-omics Analysis Reveals Different Metabolic Phenotypes Based on Molecular Characteristics in Thyroid Cancer [J]. Clin Cancer Res. 2024 Feb 16;30(4):883-894.
[3] Ringel, M. D. (2020). New Horizons: Emerging Therapies and Targets in Thyroid Cancer [J]. The Journal of Clinical Endocrinology and Metabolism, 106(1), e382.
[4] Leandro-García, L. J., & Landa, I. (2023). Mechanistic Insights of Thyroid Cancer Progression [J]. Endocrinology, 164(9), bqad118.
[5] Jung, C. K., Bychkov, A., & Kakudo, K. (2022). Update from the 2022 World Health Organization Classification of Thyroid Tumors: A Standardized Diagnostic Approach [J]. Endocrinology and Metabolism, 37(5), 703.
[6] Bychkov, A., & Jung, C. K. (2024). What’s new in thyroid pathology 2024: Updates from the new WHO classification and Bethesda system [J]. Journal of Pathology and Translational Medicine, 58(2), 98.
[7] Bai, Y., Kakudo, K., & Jung, C. K. (2020). Updates in the Pathologic Classification of Thyroid Neoplasms: A Review of the World Health Organization Classification [J]. Endocrinology and Metabolism, 35(4), 696.
[8] Basolo, F., Macerola, E., Poma, A. M., & Torregrossa, L. (2023). The 5th edition of WHO classification of tumors of endocrine organs: Changes in the diagnosis of follicular-derived thyroid carcinoma [J]. Endocrine, 80(3), 470.
[9] Khatami, F., & Tavangar, S. M. (2018). A Review of Driver Genetic Alterations in Thyroid Cancers [J]. Iranian Journal of Pathology, 13(2), 125.
[10] Xing, M. (2013). Molecular pathogenesis and mechanisms of thyroid cancer [J]. Nature Reviews. Cancer, 13(3), 184.
[11] Gill, K. S., Tassone, P., et al. (2016). Thyroid Cancer Metabolism: A Review [J]. Journal of Thyroid Disorders & Therapy, 5(1), 200.
[12] Ciavardelli, D., Bellomo, M., et all. (2017). Metabolic Alterations of Thyroid Cancer as Potential Therapeutic Targets [J]. BioMed Research International, 2017, 2545031.
[13] Coelho, R. G., Fortunato, R. S., & Carvalho, D. P. (2018). Metabolic Reprogramming in Thyroid Carcinoma [J]. Frontiers in Oncology, 8, 331588.
[14] Ju, S. H., Song, M., Lim, et al. (2024). Metabolic Reprogramming in Thyroid Cancer [J]. Endocrinology and Metabolism, 39(3), 425.
[15] Abooshahab, R., Zarkesh, M., & Hedayati, M. (2025). Metabolomics fingerprinting of thyroid malignancies: A GC/MS-based approach for subtype classification and biomarker discovery [J]. BMC Cancer, 25, 1586.
[16] Xia, J., & Zhai, B. (2026). Metabolic reprogramming orchestrates an immunosuppressive microenvironment in anaplastic thyroid cancer: Mechanisms and clinical perspectives [J]. Frontiers in Immunology, 17, 1699202.
[17] Wen, S. S., Zhang, T. T., et al. (2019). Metabolic reprogramming and its clinical application in thyroid cancer [J]. Oncology Letters, 18(2), 1579.
[18] Tiedje, V., & Fagin, J. A. (2020). Therapeutic breakthroughs for metastatic thyroid cancer [J]. Nature Reviews. Endocrinology, 16(2), 77.


Comments
Leave a Comment