Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
| Code | CSB-PA754361ESR1HU |
| Size | US$166 |
| Order now | |
| Image |
|
| Have Questions? | Leave a Message or Start an on-line Chat |
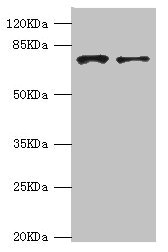
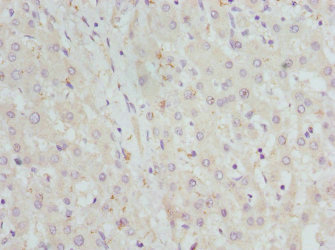
| Application | Recommended Dilution |
|---|---|
| WB | 1:1000-1:5000 |
| IHC | 1:20-1:200 |
There are currently no reviews for this product.

