Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Enzyme-linked immunosorbent assay, also known as ELISA or EIA, is a plate-based assay technique designed for detecting and quantifying substances such as peptides, proteins, antibodies and hormones by changing antibodies and color to identify a substance. In an ELISA, an antigen must be immobilized on a solid surface and then combined with an antibody that is linked to an enzyme. Detection is accomplished by assessing the conjugated enzyme activity via incubation with a substrate to produce a measureable product. The most crucial element of the detection strategy is a highly specific antibody-antigen interaction. It is a common test that detects and measure antibodies in your blood, so that an ELISA test may be used to diagnose some diseases, such as HIV-a cause of AIDS, Lyme disease, pernicious anemia, Rocky Mountain spotted fever, rotavirus squamous cell carcinoma, syphilis, et al.
CUSABIO has a sound platform for the development of ELISA kit, mature antigen-antibody research and development system. We are proficient in a variety of ELISA technologies, such as the double-antibody sandwich method, double antigen sandwich method, (direct) competition ELISA, indirect competition ELISA, blocking method, indirect ELISA and other methods. Combined with diagnostic kits development team, we are able to develop the kits with clinical diagnostic level, and make the quality in the leading place worldwide. Currently, CUSABIO now offers a broad range of ELISA kits covering over 9,000 different assay targets.
In this article, we primarily focus on the common problems of ELISA experiment.
ELISA is the abbreviation of the enzyme-linked immunosorbent assay used to identify the presence of specific proteins and to determine their concentrations. There are four types of ELISA including sandwich, competitive, indirect and direct ELISA. The common procedures are Storage, Reagents preparation, ELISA plate, Samples or reagents adding, Incubation, Washing and Reading. Please refer to our ELISA protocol for details. As shown in Figure 1, there are five procedures of ELISA in brief.
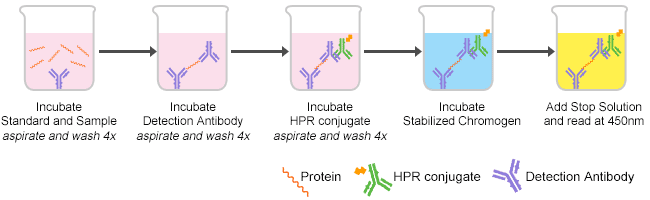
Figure 1. The procedures of ELISA
The ELISA results are generally based on the color depth of the chromogenic substrate. The color reaction is required to be carried out at 37 °C for about 10 minutes, then the color reaction is terminated with a stop solution, and the absorbance at a specific wavelength is monitored with a microplate reader. Since the detection path of each reaction well is perpendicular to the microplate reader, the bottom of each reaction well should be kept clean when testing, and the amount of chromogenic substrate and stop solution should be accurate, because the inside of the well The amount of liquid will affect the final degree. In this part, we summarize several results of the color reaction in ELISA experiment. Besides that, as shown in Table 1, we also conclude the causes and solution, respectively.
Table 1. The problems of the color reaction
| cause | solution |
|---|---|
| Assay set up incorrectly, used incorrect reagents or Incorrect wavelength | Review protocol. Repeat assay using a positive control and check plate reader for wavelength, filters, gain etc. |
| Not enough antibody used | Try different concentration of the primary and/or secondary antibody. |
| Incubation time too short | Incubate samples overnight at 4°C or follow the manufacturer guidelines. |
| Antibody stored at 4°C for several weeks or subjected to repeated freeze/thaw cycles | Use a fresh aliquot of antibody that has been stored at -20°C or below. |
| Recognition of epitope impeded by adsorption to plate | To enhance detection of a peptide by direct or indirect ELISA, conjugate peptide to a large carrier protein before coating onto the microtiter plate. |
| Slow color development of enzymatic reaction | Prepare substrate solution immediately before use. Ensure the stock solution has not expired and is not contaminated. Allow longer incubation. |
| cause | solution |
|---|---|
| Improper storage of ELISA kit | Store all reagents as recommended. Please note that all reagents may not have identical storage requirements. |
| Plate reader settings incorrect | Check plate reader for wavelength, filters, gain, etc. |
| Inactive detection reagent or detection reagent too dilute | Ensure reporter enzyme/flour has the expected activity, or use a higher concentration of detection reagent. |
| Insufficient amount of antigen was coated to microtiter plate | Use more antigen for coating or very coating buffer. |
| Not enough antibody used | Increase concentration of the primary and/or secondary antibody. Optimize antibody concentrations for your assay. |
| Mixing or substituting reagents from different kits | Avoid mixing components from different kits. |
| Incubation temperature too low | Optimize the incubation temperature for your assay. Reagents should be at room temperature before beginning the assay. |
| Assay plates were compromised or previously used. | Be sure to refrigerate plates in sealed bags with a desiccant to maintain stability. Prevent condensation from forming on plates by allowing them to equilibrate to room temperature while in the packaging. If partial plates are used, you must be sure to label used wells to prevent reuse; cover them with sealing tape and use the remaining wells as soon as possible. Do not store partially used plates with other plates. Include a desiccant in the storage bag. |
| cause | solution |
|---|---|
| Too much antibody used | Optimize antibody concentrations for your assay with different dilutions. |
| Too much detection reagent | Ensure the reagent has been diluted properly or repeat assay with a higher dilution of detection reagent. |
| Incubation temperature too high | Try different temperature for optimizing your assay. |
| Reaction not stopped | Use stop solution to prevent overdevelopment. |
| Waiting too long to read plate after adding stop solution | Read plate immediately after adding stop solution. |
| Incubation with substrate carried out in the light | Perform substrate incubation in the dark. |
| Non-specific binding of antibody | Use a suitable blocking buffer or use an affinity-purified antibody. |
| Dirty plate | Clean the plate bottom carefully and reread. |
| cause | solution |
|---|---|
| Incubation temperature is wrong | Ensure plates and reagents are kept at room temperature. |
| Contaminated solutions | Make fresh solutions. |
| Detection reagent too old, contaminated or used at the wrong pH | Use fresh detection reagents at the correct pH. |
| Wrong conjugate was used, conjugate was prepared incorrectly or has deteriorated. | Be sure that the conjugate used is the one that came with the kit. All conjugates are kit- and lot-specific. If preparation of a working conjugate is needed, be sure that the concentrate and diluent are mixed in correct volumes. Do not prepare the working solution too far in advance and do not save any unused portion for future use. If no conjugate preparation is necessary, be sure to pour out only the amount required for immediate use and do not return any unused portion to the stock bottle. |
| Wash buffer contains sodium azide. | Avoid sodium azide in the wash buffer. |
There are numerous problem in ELISA experiment except for abnormal color reaction, involving standard curve, data analysis, the repeatability of experiment, et al. In this part, we list a few representative questions as follows, if you want to know more, please click the below link: https://www.cusabio.com/m-307.html.
In the ELISA data analysis, the standard curve is the most important factor for tested samples. Therefore, how to make a fine standard curve and the quality of the standard are critical. Here, we conclude several reasons and solutions about the question that Why does the standard curve look poorly as follows. If you want to obtain the way of making standard curve, please click here.
a. Standard solution is Improper, you should confirm dilutions and make them correctly.
b. Standard is reconstituted improperly. You should spin vial briefly before opening and inspect for undissolved material after reconstituting.
c. Standard is degraded, you should store and handle standard as recommended.
d. Curve doesn't fit scale, please try plotting using different scales e.g. log-log, 5 parameter logistic curve fit.
e. Pipetting error, Using calibrated pipettes and proper pipetting technique to revise.
Wrong filter is used when taking readings. Wavelength should be 450nm with a 650nm wavelength correction for TMB. Checking plate reader for wavelength, filters, gain etc. again before reading.
a. Improper storage of the kit or poor storage environment. You should store all components as recommended on data sheet rather than room temperature for excess time.
b. Incorrect preparation of standard. You should reconstitute standard strictly with the recommended diluent according to the kit protocol. You should prepare reagents in 10 minutes prior to use and add them to wells promptly.
c. Insufficient mixing after adding samples. You should fully mix reagents in the vortex mixer when adding several reagents at the same time. Be careful when holding reagents to avoid splashing.
d. Poor repeatability of the plate readings. You should calibrate the plate reader.
e. Inconsistent incubation time, washing condition, color development condition and operators. You should repeat the assay of standard. Ensure consistent reactivity condition and operators.
f. Improper washing. You should add 200μL of wash buffer or fully fill into every well with pipette but no overflows are allowed. Check that all ports of the plate washer are unobstructed to ensure sufficient washing.
g. Uneven temperature. You should keep constant temperature during incubation to avoid temperature fluctuations.
h. Too much residual on the wall of the wells when adding or the bottom of wells scratched with pipette tip. You should lower the pipette tips along the wall of wells when adding slowly and carefully. Do not touch the bottom of wells.
i. Reused materials. You should change pipette tips between samples and reservoirs between reagents.
j. Occasional positive and negative values close to the cut off value. You should set 3 duplicates for the same sample and the same result over 2 samples.
k. Cross contamination when adding samples. You should avoid cross contamination when adding samples.
a. Cross contamination during manual washing. You should reduce the cross contamination by promptly removing the liquid in wells after 3 times of filling washing buffer during manual washing and then setting the soak time the next times.
b. Cross contamination when patting the plate. You should use proper paper towels when patting the plate. Do not bring unrelated materials into the plate. Do not pat at the same place to avoid cross contamination.
c. Contamination due to long storage of samples. You should keep samples fresh or store them under low temperate to avoid contamination.
d. Abnormal developed color due to insufficient filling or too much residual when the plate washer is obstructed. You should fully fill into every well with pipette but no overflows are allowed. Check that all ports of the plate washer are unobstructed to ensure sufficient washing.
e. Coagulationor interference of precipitates or residual cell caused by incomplete centrifugation of samples. You should complete centrifugation of serum and plasma.
f. Wrong preparation of wash buffer or misuse of concentrated wash buffer. You should prepare wash buffer as manual required.
Besides that, a sweet guy in our company also writes an article about Pre-experiment. If you want to see the full article, you can click the following link to enter the site: https://www.cusabio.com/m-226.html.


Comments
Leave a Comment