Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
The quest to harness the body's immune system to fight disease took a monumental leap forward with the development of phage display technology. Pioneered by George Smith in 1985 with the display of peptides on filamentous phage surfaces [1], and later revolutionized by Sir Gregory Winter and colleagues for antibody engineering, phage display earned its creators the Nobel Prize in 2018 for the phage display of peptides and antibodies. From its origins as a novel screening tool, phage display has become the cornerstone of modern therapeutic biologic discovery, transforming the development of antibodies and beyond.
This article will move from basic principles to library design, selection workflows, and the path from initial hits to drug candidates, with an eye on practical implications for therapeutic development.
Table of Contents
2. Fundamentals of Phage Display Technology
3. Phage Display for Antibody Discovery
4. From Hits to Leads: Characterization and Engineering
5. Therapeutic Impact of Phage Display–Derived Antibodies
6. Broader Applications of Phage Display
7. Technical Advances and Future Directions of Phage Display
Phage display is a molecular technique that presents peptides or proteins on the surface of bacteriophages while carrying the encoding DNA inside the same particle. This physical connection between genotype (a DNA sequence that encodes the displayed molecule) and phenotype (the displayed molecule) enables direct selection of binding functions and immediate recovery of the corresponding sequences [1-3]. Since the establishment of the conceptual foundation of phage display by George Smith in 1985, it has become an important platform for studying protein–protein, protein–peptide, and protein–DNA interactions and for discovering therapeutic antibodies and peptides.
Over the past three decades, this approach has become central to therapeutic antibody discovery, with multiple approved monoclonal antibodies (mAbs) and many clinical candidates derived from phage display campaigns. For teams working on new biologics, it now sits alongside transgenic animals and B-cell cloning as a core discovery platform.
In a typical phage display construct, a gene encoding a peptide, protein domain, or antibody fragment is fused in-frame to a phage coat protein gene so that the resulting fusion is incorporated into the virion envelope. The same virion packages the corresponding DNA, which means that binding properties observed at the surface can be traced back to their exact sequences [2,3].
These "fusion phage" can be enriched by affinity selection ("biopanning") against immobilized ligands in an in vitro process analogous to natural selection. This makes phage display a powerful platform for building and screening very large libraries of variants to identify rare high‑affinity binders [2-4].
While filamentous phages are the mainstay for antibody phage display, several lytic phages have been adapted as alternative display platforms, including T4, T7, and λ phages.
E.coli filamentous bacteriophages (f1, fd, M13) are the most widely used vectors for antibody phage display. They are rod‑shaped viruses with single‑stranded DNA genomes and a flexible, non‑lytic life cycle in Escherichia coli, which facilitates library amplification without host cell lysis. M13, in particular, has become the standard, with a cylindrical particle composed of thousands of copies of the major coat protein pVIII and a few copies of minor coat proteins, including pIII, pVI, pVII, and pIX [2,4,5].
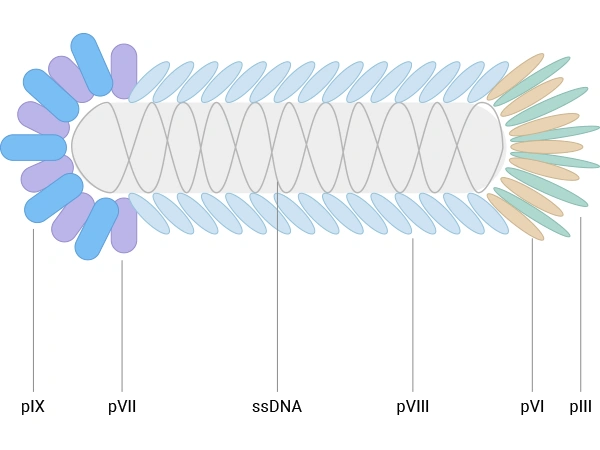
Figure 1. The schematic diagram of E. coli filamentous bacteriophage
Most antibody and peptide display constructs use either:
Other coat proteins, such as pVI, pVII, and pIX, have also been explored as alternative scaffolds and can complement pIII/pVIII systems, particularly for cDNA libraries or high‑density peptide display. Nevertheless, for therapeutic antibody discovery, M13‑based pIII and pVIII display systems remain dominant in both academic and industrial settings [2-4].
T4 phage is a bacteriophage that infects Escherichia coli and belongs to the T-even phage group. It has a large structure (~90 nm wide, 200 nm long) and a 169 kbp double-stranded DNA genome encoding 289 proteins. Its capsid is composed of three essential proteins: gp23 (forms the hexagonal lattice), gp24 (forms pentamers at eleven vertices), and gp20 (forms the dodecameric portal vertex for DNA entry/exit). Additionally, the capsid is decorated with two non-essential outer proteins: HOC (highly antigenic outer capsid protein) and SOC (small outer capsid protein).
T4 phage can display peptides and proteins via nonessential outer capsid proteins such as SOC and HOC at high density, which is advantageous for immunogenic presentation and cDNA library expression [7,8].
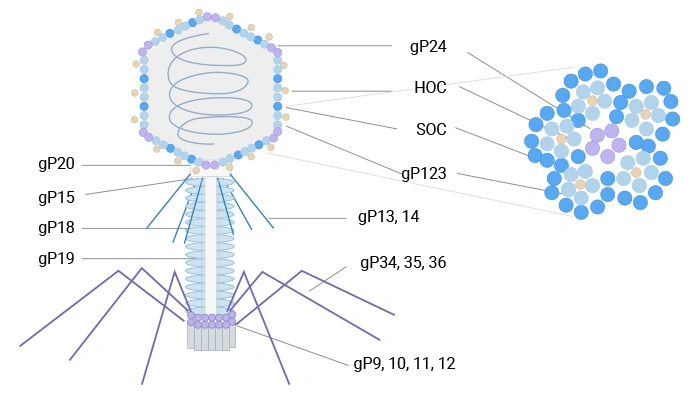
Figure 2. The schematic diagram of T4 phage
T7 phage, an icosahedral podovirus, contains a linear double-stranded DNA genome within a head-tail structure. Its capsid comprises 415 copies of the major coat protein gp10, forming 60 hexamers and 11 pentamers. Two naturally occurring isoforms, gp10A and gp10B (in a ~9:1 ratio), arise from a translational frameshift; gp10B contains an additional 52 C-terminal residues. Fusion proteins are displayed at this extended C-terminus of gp10B, minimizing steric interference. T7 phage exhibits high stability under extreme temperature and pH conditions, facilitating robust high-throughput affinity selection.
T7 phage uses its capsid protein gp10B to display fusion partners and supports display of relatively large protein fragments at low copy number, benefiting from high stability and rapid lytic growth; it has proven useful in antigen discovery, protein interaction studies, and cancer‑related profiling.
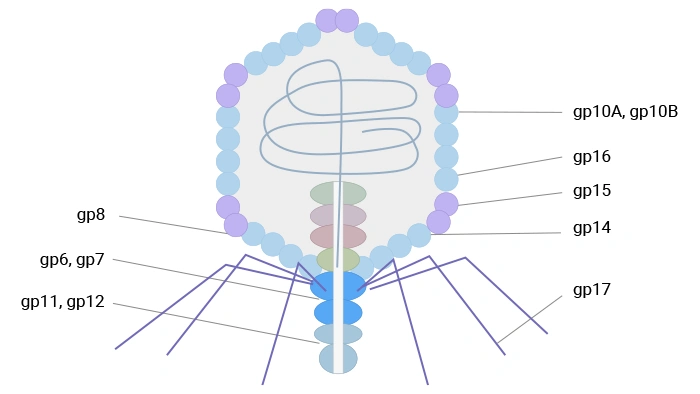
Figure 3. The schematic diagram of T7 phage
Lambda (λ) phage is a bacterial virus that infects Escherichia coli. It features an icosahedral head, primarily composed of the major coat protein gpE (~415 copies) and stabilized by the decoration protein gpD (~402-420 copies). The head connects to a flexible tail built from the major tail protein gpV. Both gpV and gpD have been exploited for phage display: initially, a low-display system was developed for C-terminal fusions to gpV, suitable for selecting high-affinity binders. Later, more efficient systems were engineered for N- or C-terminal display on gpD, enabling the multivalent presentation of peptides or large protein domains and making λ phage a robust platform for screening diverse libraries.
λ phage has also been engineered for display on capsid protein gpD or tail protein gpV, allowing multivalent display of peptides or protein domains for library screening. These lytic systems broaden the range of targets and applications but, in the context of therapeutic antibody discovery, are generally used as complementary tools alongside filamentous M13‑based systems [2,7,8,9].
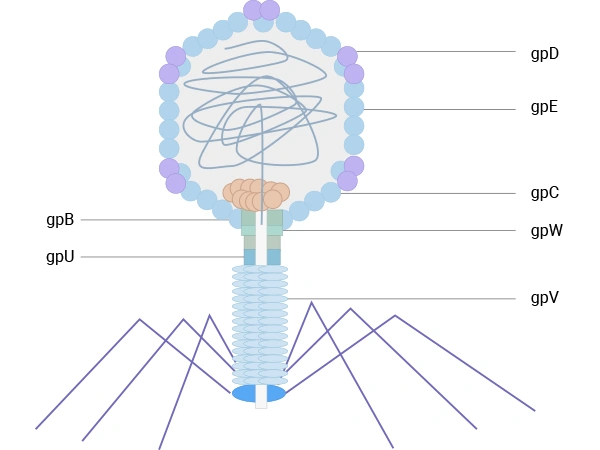
Figure 4. The schematic diagram of λ phage
Although phage display began with short peptides, it has expanded to many types of binding domains [10]. Commonly displayed entities include:
The antibody phage display workflow initiates with antibody-library construction, proceeds with ligating the variable heavy (VH) and variable light (VL) PCR products into a phage display vector, and ends with the analysis of monoclonal antibody (mAb) clones.
A library is screened for phage binding to an antigen through its expressed surface mAb via (bio-)panning. A standard phage display selection (biopanning) can be summarized in five core steps:
① Library construction – Antibody or peptide genes are cloned into phage or phagemid vectors so that each phage displays a single variant on its surface (typically via pIII or pVIII), and the collection of distinct clones forms a large, diverse library [2,3].
② Binding – The library is incubated with an immobilized target, such as a purified protein, a cell‑surface antigen, or live cells and tissues, allowing specific binders to attach [2,3,11].
③ Washing – Non‑binding and weakly binding phages are removed by washing under defined stringency conditions, thereby enriching particles with higher affinity for the target [2,3].
④ Elution – Bound phage is recovered by elution, for example, using pH changes, competitive ligands, or protease treatment [3].
⑤ Amplification – Eluted phage infects fresh E. coli host cells and are amplified, generating an enriched pool that can undergo additional rounds of selection to further focus on the best binders [2,3].
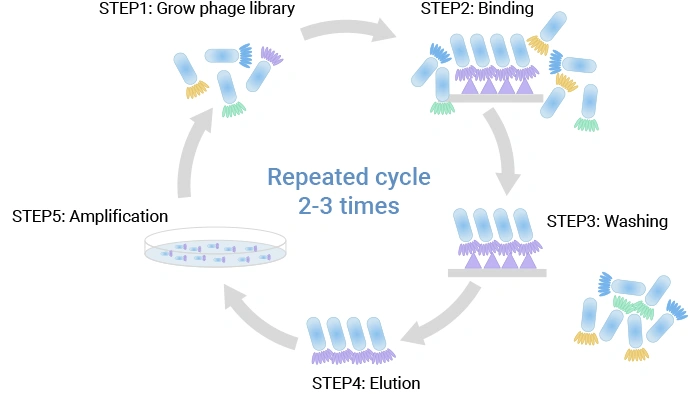
Figure 5. The phage display protocol
Typically, 2–3 rounds of this cycle are performed, with progressively tighter washing and lower antigen concentration to drive selection toward higher affinity and specificity. Cell‑based panning, where phages are incubated with living cells expressing the target, is especially useful for membrane proteins, including many drug targets in oncology [11].
Antibody phage display libraries are collections of phage that together present a very large and diverse set of antibody fragments. The main library types are:
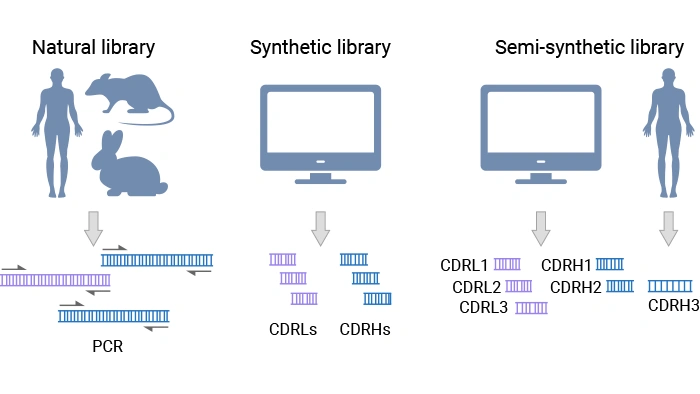
Figure 6. Types of antibody phage display libraries [2]
Immune libraries often provide higher‑affinity hits for the immunizing antigen. Still, they are narrower in scope, whereas naïve and synthetic libraries offer broader coverage and are well-suited for targets where immunization is difficult or not possible [2,3].
Early phage display libraries were limited in diversity and used simple or poorly controlled CDR (complementary determining region) designs. Over time, second‑ and third‑generation libraries have incorporated:
These improvements have led to more reliable discovery of high‑affinity, drug‑like antibodies directly from a single library platform [2].
Different fragment formats are used in antibody phage display, each with pros and cons [2,10].
Format choice affects display efficiency, stability during panning, and the ease of converting hits into full‑length IgG.
Because phage display selections are fully in vitro, you can tune the conditions to favor particular antibody properties.
To enrich for high affinity, researchers often use off‑rate selections, where bound phages are subjected to long washes or competitor molecules so that only slow‑dissociating clones remain. For specificity, negative selection against related proteins or cell lines helps remove cross‑reactive clones [2].
Selections can also be designed around function, such as pH‑dependent binding for recycling antibodies, or promoting receptor internalization for antibody–drug conjugate (ADC) delivery. These functional selection schemes move phage display beyond simple binding toward more pharmacologically relevant behavior [2].
Next‑generation sequencing (NGS) has become a powerful companion to phage display. By sequencing antibody genes from each round of panning, teams can:
These deep profiles support more informed hit picking and iterative optimization of selection strategies. Combined with computational models, NGS data can also help map sequence–function relationships, guiding the design of new libraries or rational diversification around a lead clone [12].
Antibody phage display libraries are powerful in vitro platforms that rapidly screen billions of antibody variants for binders against virtually any target. By capturing diverse antibody repertoires—from immunized, naïve, or synthetically designed sources—these libraries enable the direct discovery of high-affinity fragments like scFvs, Fabs, and VHHs, which are foundational for developing research reagents and therapeutic antibodies.
The critical phase of antibody discovery transitions from library screening to rigorous hit validation and optimization. This process involves confirming antigen-specific binding through techniques like ELISA and SPR, followed by functional characterization in disease-relevant assays. Lead candidates often undergo further engineering—such as affinity maturation and developability optimization—to enhance their therapeutic potential, ultimately paving the way for converting fragments into full-length biologic drugs.
After panning, individual clones are isolated and screened. Initial screens often use:
Secondary assays then probe key properties such as affinity, specificity, and cross‑reactivity, using techniques like ELISA, surface plasmon resonance (SPR), biolayer interferometry (BLI), or flow cytometry [3,10].
For therapeutic candidates, functional assays are essential. Depending on the target, these might measure neutralization of a ligand, receptor activation or blockade, or effector functions such as antibody‑dependent cellular cytotoxicity (ADCC). At this stage, many programs prioritize antibodies that combine strong binding with robust performance in disease‑relevant cell‑based assays [10,11].
Often, initial antibodies from phage display benefit from further optimization. Affinity maturation commonly uses the following libraries:
These refined libraries go through additional rounds of panning under more stringent conditions to obtain higher‑affinity binders [2,11].
In parallel, antibodies are engineered to improve “developability” — their behavior as drug candidates. This can involve removing aggregation‑prone motifs, reducing chemical liabilities such as deamidation hotspots, and minimizing predicted immunogenicity. Modern synthetic human libraries increasingly bake these considerations into their initial design, reducing the need for extensive post‑hoc engineering [2,10].
Once a suitable fragment is identified, it is usually converted to a full‑length IgG or alternative therapeutic format, such as an Fc‑fusion. Fc engineering can then tune half‑life and effector function, completing the path from phage‑displayed fragment to a clinically relevant molecule [10].
Phage display has revolutionized therapeutic antibody discovery, generating numerous approved drugs for cancer, autoimmune, and infectious diseases. This powerful platform enables rapid isolation of fully human antibodies without animal immunization, while its target-agnostic nature allows quick adaptation to emerging therapeutic needs. Discover why this technology offers a unique combination of speed, control, and versatility for modern drug development.
Phage display has already produced more than a dozen approved therapeutic antibodies and many more in clinical trials, covering indications such as cancer, autoimmune diseases, and infectious diseases. A key strength is the ability to isolate fully human antibodies without immunizing animals, which can simplify development and reduce immunogenicity concerns [2,3,10].
Because the platform is target‑agnostic, the same libraries and workflows can be applied to oncology, inflammatory diseases, and emerging pathogens, making phage display highly adaptable to changing therapeutic needs [2,10].
Compared with traditional in vivo immunization, phage display offers several practical advantages [2,3]:
Rather than replacing animal‑based methods outright, phage display complements them and often serves as a rapid route to backup series or differentiated antibodies against the same target [2].
While therapeutic antibody discovery remains a flagship application, phage display technology demonstrates remarkable versatility that extends far into basic and translational research. By screening diverse peptide or protein libraries, this platform enables critical advancements in epitope mapping, ligand-receptor identification, protein interaction analysis, and the direct evolution of bioactive molecules. These broader applications solidify phage display as an indispensable and powerful tool for tackling complex biological questions and developing novel diagnostic and therapeutic strategies.
Phage display supports many applications beyond therapeutic antibody discovery.
These broader uses highlight why phage display remains a versatile platform for both basic research and translational development.
Phage display technology is evolving beyond simple selection into a predictive, data-driven discovery engine. The integration of next-generation sequencing and artificial intelligence is revolutionizing library design and hit optimization, while hybrid approaches that combine phage display with other screening modalities are tackling increasingly complex targets. These advancements, coupled with fully automated high-throughput workflows, are poised to significantly accelerate the development of next-generation biotherapeutics.
Recent phage display libraries are designed using insights from NGS, structural biology, and developability rules. Diversity is focused on CDR positions that contribute most to binding, while disfavored amino acids and unstable motifs are excluded. This “developability‑aware” design improves the odds that hits will express well and behave like drug candidates [2].
Machine learning models trained on large antibody datasets are now being explored to predict affinity, specificity, and liabilities from sequence, helping to rank hits and suggest beneficial mutations. As these tools mature, they are likely to further streamline phage display workflows [12].
Phage display is increasingly used alongside other discovery methods. For example:
These hybrid approaches combine the strengths of different technologies to address challenging targets more effectively.
On the process side, robotics, high‑throughput screening, and standardized analytics have turned phage display into a scalable industrial platform. Automated panning, parallel campaigns against panels of antigens, and integrated data pipelines now allow discovery teams to run many projects in parallel while maintaining consistent quality [12].
At the same time, antigen presentation formats are improving—from more native membrane protein displays to better control of antigen orientation—which helps align in vitro binding with in vivo performance [11].
Q1: Why can't I find a blue plaque after the second round of phage amplification, with a plaque titer assay?
A1: There could be several reasons as follows:
a. Please confirm if the dilution ratio is correct.
b. There are other phage contaminations in the amplification process, and the bacteria are lysed.
c. The phage has not been centrifuged when the precipitation is insufficient.
d. The X-gal is a failure.
Q2: While assaying the titer of phage, why is the plate all blue?
A2: Maybe the titer of phage is too high; you should dilute more gradients. And please control the incubation time, so that the bacterial plaques do not become too large to be confluent.
Q3: Antibody Selection from phage display Libraries: after panning and dilution, why is there no plaque on the plate?
A3: There could be several reasons as follows:
a. The medium is not suitable; maybe you have added the wrong antibiotic.
b. The dilution ratio of the phage is too large. In general, after panning, the eluted phage titer should be 105-7.
c. The phage is not neutralized after elution, so it is inactivated.
d. The incubation temperature is not suitable, so the flagella of bacteria has not been expressed.
e. Please confirm that if the host bacteria are correct, they can be infected and express flagella.
Q4:How can I manipulate the plaque size of bacteriophage?
A4: The size of the plaque is a trait of the replication cycle. If the bacterial plaques are becoming confluent, you could try to do something to decrease the rate at which they grow. Such as follows:
a. Removing nutrients may reduce the burst size or reduce the number of subsequent infections, and subsequently produce smaller plaques.
b. Reduce temperature, or shorter incubation times.
c. Reduce the multiplicity of infection.
d. Try adding more quantity and a higher concentration of host cells.
Q5: After screening for several rounds, why did I obtain the positive clones with the same sequences?
A5: There could be several reasons as follows:
a. The capacity of your antibody library is too small, and the diversity is poor. You can try screening with multiple libraries.
b. The washing condition is too strict. You can try to reduce the concentration of detergent in the washing buffer appropriately.
c. The washing time is too excessive, resulting in the loss of most of the clones. You can try to reduce washing times appropriately.
d. The panning round is excessive, and you can select and check the clone after the previous screening.
e. Please determine if the phage has a droplet of 100 times its capacity.
Q6: After screening, why are there no positive clones obtained by the ELISA assay?
A6: There could be several reasons as follows:
a. During the screening process, the non-specific conjugated phages are excessive, and the phages adsorbed by the antigen are few. So that there are no positive clones that could be selected.
b. The antibody has not been expressed, so we could determine it by Western blot.
c. The step of the ELISA assay is not correct, so we could determine it by positive control.
d. The single-plaque plate has been contaminated, and the E.coli with the same resistance has been selected.
Q7: Why can't screened antibodies recognize endogenous proteins?
A7: There could be several reasons as follows:
a. The antibodies that have been screened are not specific for target proteins, but specific for the vector or label sequence.
b. The expression of endogenous protein is too low to be detected.
c. Low antibody affinity.
Phage display has evolved from a clever molecular trick into a mature, widely adopted engine for therapeutic antibody and peptide discovery. By coupling massive library diversity with controllable in vitro selection, it offers a powerful and flexible route from target concept to high‑quality antibody leads.
Ongoing advances in library design, sequencing, computation, and automation are keeping phage display at the center of biologics discovery, especially for difficult targets and tailored antibody functions. For researchers entering the field today, understanding phage display is no longer optional—it is part of the basic toolkit for turning molecular ideas into real therapeutic candidates.
References
[1] Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface [J]. Science. 1985 Jun 14;228(4705):1315-7.
[2] Zhang, Y. (2023). Evolution of phage display libraries for therapeutic antibody discovery [J]. MAbs, 15(1), 2213793.
[3] Z Chan, C. E., C Lim, A. P., MacAry, P. A., & Hanson, B. J. (2014). The role of phage display in therapeutic antibody discovery [J]. International Immunology, 26(12), 649.
[4] Tikunova, N., & Morozova, V. (2009). Phage Display on the Base of Filamentous Bacteriophages: Application for Recombinant Antibodies Selection [J]. Acta Naturae, 1(3), 20.
[5] Rakonjac J, Model P. Roles of pIII in filamentous phage assembly [J]. J Mol Biol. 1998 Sep 11;282(1):25-41.
[6] Fagerlund A, Myrset AH, Kulseth MA. Construction and characterization of a 9-mer phage display pVIII-library with regulated peptide density [J]. Appl Microbiol Biotechnol. 2008 Oct;80(5):925-36.
[7] Slootweg, E. J., Keller, H. J., et al. (2006). Fluorescent T7 display phages obtained by translational frameshift [J]. Nucleic Acids Research, 34(20), e137.
[8] Deng X, Wang L, You X, Dai P, Zeng Y. Advances in the T7 phage display system (Review) [J]. Mol Med Rep. 2018 Jan;17(1):714-720.
[9] Kalniņa Z, Siliņa K, et al. Evaluation of T7 and lambda phage display systems for survey of autoantibody profiles in cancer patients [J]. J Immunol Methods. 2008 May 20;334(1-2):37-50.
[10] Alfaleh, M. A., Alsaab, H. O., et al. (2020). Phage Display Derived Monoclonal Antibodies: From Bench to Bedside [J]. Frontiers in Immunology, 11, 567223.
[11] Istomina PV, Gorchakov AA, Paoin C, Yamabhai M. Phage display for discovery of anticancer antibodies [J]. N Biotechnol. 2024 Nov 25;83:205-218.
[12] Huang, J., Takakusagi, Y., & Ru, B. (2022). Editorial: Phage display: Technique and applications [J]. Frontiers in Microbiology, 13, 1097661.


Comments
Leave a Comment