Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Fundamental research, also called pure research or basic research, advances fundamental knowledge about the world. It aims to improve scientific theories for improved understanding or prediction of natural or other phenomena. Pure research is the source of most new scientific ideas and ways of thinking about the world. It can be exploratory, descriptive, or explanatory; however, explanatory research is the most common. Here, we primarily focus on basic research of molecular biology.
Molecular biology is a branch of biology that concerns the molecular basis of biological activity between biomolecules in the various systems of a cell, including the interactions between DNA, RNA, proteins and their biosynthesis, as well as the regulation of these interactions. The main principle of molecular biology is genetic central dogma, as shown in the figure 1. Here, we put several databases of molecular biology research, primarily including gene information, gene expression information, gene function analysis, transcription factor, miRNA analysis and prediction, and others.
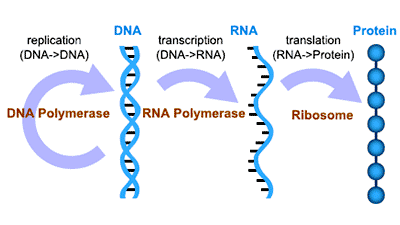
Figure 1. The brief diagram of genetic central dogma
A gene is a locus (or region) of DNA that encodes a functional RNA or protein product, and is the molecular unit of heredity. Compared with DNA, The DNA contains large sequences that do not code for any protein and their function is not known. In another words, DNA is larger than gene. There are nearly 50,000 and 100,000 genes with each being made up of hundreds of thousands of chemical bases. The gene information plays a crucial role in transcription and translation. Here, we list three powerful databases for looking up gene information.
GeneCards (https://www.genecards.org/) is a searchable, integrative database of human genes that provides comprehensive, user-friendly information on all annotated and predicted human genes. It automatically integrates gene-centric data from ~150 web sources, including genomic, transcripts, aliases, disorders, domains, drugs, expression, and localization, et al.
BioGPS (http://biogps.org/#goto=welcome) is a free extensible and customizable gene annotation portal based on a loose federation of existing genetic and genomic resources. BioGPS allows users to easily explore the landscape of gene annotation resources for one or more genes of interest. Currently BioGPS focuses on annotation for human, mouse, and rat genes.
Moreover, BioGPS also emphasizes two key design features. Firstly, BioGPS is based on a simple, unstructured plugin interface that allows for simple community extensibility. Secondly, BioGPS also implements a powerful user interface that enables precise user customizability. In sum, these two design principles enable BioGPS to harness the principle of community intelligence toward the goal of efficiently organizing and querying online gene annotation resources.
The UCSC Genome Browser (http://genome.ucsc.edu/) is an on-line, and downloadable, genome browser hosted by the University of California, Santa Cruz (UCSC). It is an interactive website offering access to genome sequence data from a variety of vertebrate and invertebrate species and major model organisms, integrated with a large collection of aligned annotations. The Browser is a graphical viewer optimized to support fast interactive performance and is an open-source, web-based tool suite built on top of a MySQL database for rapid visualization, examination, and querying of the data at many levels.
Gene expression is the process by which information from a gene is used in the synthesis of a functional gene product. These products are often proteins, which go on to perform essential functions as enzymes, hormones and receptors, for example. But in non-protein coding genes such as transfer RNA (tRNA) or small nuclear RNA (snRNA) genes, the product is a functional RNA.
Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/), also known as GEO, is a public functional genomics data repository supporting MIAME-compliant data submissions. Array- and sequence-based data are accepted. Tools are provided to help users query and download experiments and curated gene expression profiles.
ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) is one of the repositories recommended by major scientific journals and aims to archive functional genomics data from microarray and sequencing platforms to support reproducible research. To serve this mission, we facilitate submissions in compliance with Minimum Information About a Microarray Experiment (MIAME) and Minimum Information About a Sequencing Experiment (MINSEQE) guidelines.
The Cancer Genome Atlas (https://www.cusabio.com/c-20839.html), also known as TCGA, is a project collaboratively monitored by the National Cancer Institute (NCI) and the National Human Genome Research Institute in the United States (NHGRI) that has generated comprehensive, multi-dimensional maps of the key genomic changes in 33 types of cancer. It aims to apply high-throughput Genomic analysis techniques to help people have a better understanding of cancer, thereby improving the prevention, diagnosis, and treatment of cancer. For more information about TCGA, you can click the following link to view: https://www.cusabio.com/c-20839.html.
Gene Function Analysis is an important method to analyse the function of a series of unknown genes based on genomic studies.
The Database for Annotation, Visualization and Integrated Discovery (https://david.ncifcrf.gov/), also known as DAVID, comprises a full Knowledgebase update to the sixth version of our original web-accessible programs. DAVID now provides a comprehensive set of functional annotation tools for investigators to understand biological meaning behind large list of genes. For any given gene list, DAVID tools are able to:
Metascape (http://metascape.org/gp/index.html#/main/step1) is a free gene annotation and analysis resource that helps biologists make sense of one or multiple gene lists. Metascape provides automated meta-analysis tools to understand common or unique pathways and protein networks within a group of orthogonal target-discovery studies. Note, all analysis results are presented in a web report, which contains Excel annotation and enrichment sheets, PowerPoint slides, and custom analysis files for further offline analysis or processing.
Additionally, in this page, several sweet guys from Metascape list a series of Frequently Asked Questions about this online analysis tool, involving how to cite Metascape, what species does Metascape support and what type of gene identifiers are supported, et al.
XTalkDB (http://www.xtalkdb.org/home) is a database of signaling pathway crosstalk. As you knows, analysis of signaling pathways and their crosstalk is a cornerstone of systems biology. Before XTalkDB developed, there was no database that carefully and explicitly documents crosstalk between specific pairs of signaling pathways.
After unremitting efforts, several excellent researchers have developed XTalkDB (http://www.xtalkdb.org) to fill this very important gap. XTalkDB contains curated information for 650 pairs of pathways from over 1600 publications. In addition, the database reports the molecular components (e.g. proteins, hormones, microRNAs) that mediate crosstalk between a pair of pathways and the species and tissue in which the crosstalk was observed. The XTalkDB website provides an easy-to-use interface for scientists to browse crosstalk information by querying one or more pathways or molecules of interest.
Transcription factor, also called sequence-specific DNA-binding factor, is a protein that controls the activity of a gene by determining whether the gene’s DNA is transcribed into RNA. The enzyme RNA polymerase catalyzes the chemical reactions that synthesize RNA, using the gene’s DNA as a template. Transcription factors are of clinical significance for at least two reasons: mutations can be associated with specific diseases, and they can be targets of medications. Here, we introduce two useful databases for transcription factor research.
A iRegulon (http://iregulon.aertslab.org/) consists of a transcription factor (TF) and its direct transcriptional targets, which contain common TF binding sites in their cis-regulatory control elements. The iRegulon plugin allows you to identify regulons using motif and track discovery in an existing network or in a set of co-regulated genes.
Transcription Factor Checkpoint (http://www.tfcheckpoint.org/) is a resource for Human, Mouse and Rat Transcription Factors. The Transcription Factors (TFs) in TFcheckpoint are manually checked for experimental evidence supporting their role in regulation of RNA polymerase II and specific DNA binding activity.
miRNA, also called microRNA, is a small non-coding RNA molecule (containing about 22 nucleotides) found in plants, animals and some viruses, that functions in RNA silencing and post-transcriptional regulation of gene expression. The function of miRNAs appears to be in gene regulation via base-pairing with complementary sequences within mRNA molecules. For that purpose, a miRNA is complementary to a part of one or more messenger RNAs (mRNAs).
Animal miRNAs are usually complementary to a site in the 3' UTR whereas plant miRNAs are usually complementary to coding regions of mRNAs. In fundamental research, miRNAs is a common useful method to down-regulate target gene expression. In this part, we collect some powerful miRNA analysis and prediction tools, as follows:
starBase (http://starbase.sysu.edu.cn/) is an open-source platform for studying the miRNA-ncRNA, miRNA-mRNA, ncRNA-RNA, RNA-RNA, RBP-ncRNA and RBP-mRNA interactions from CLIP-seq, degradome-seq and RNA-RNA interactome data. Currently, starBase identifies more than 1.1 million miRNA-ncRNA, 2.5 million miRNA-mRNA, 2.1 million RBP-RNA and 1.5 million RNA-RNA interactions from multi-dimensional sequencing data.

miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/php/index.php) is the experimentally validated microRNA-target interactions database. As a database, miRTarBase has accumulated more than three hundred and sixty thousand miRNA-target interactions (MTIs), which are collected by manually surveying pertinent literature after NLP of the text systematically to filter research articles related to functional studies of miRNAs.
miRWalk (http://mirwalk.umm.uni-heidelberg.de/), a comprehensive database that provides predicted as well as validated miRNA binding site information on miRNAs for human, mouse and rat. miRWalk gathers all the information on predicted as well as validated miRNA targets, this database has the potential of becoming an important resource for scientists engaged with miRNA research.
The miRBase database (http://www.mirbase.org/) is a searchable database of published miRNA sequences and annotation. Each entry in the miRBase Sequence database represents a predicted hairpin portion of a miRNA transcript (termed mir in the database), with information on the location and sequence of the mature miRNA sequence (termed miR). Both hairpin and mature sequences are available for searching and browsing, and entries can also be retrieved by name, keyword, references and annotation. All sequence and annotation data are also available for download.
TargetScan (http://www.targetscan.org/vert_72/) predicts biological targets of miRNAs by searching for the presence of conserved 8mer, 7mer, and 6mer sites that match the seed region of each miRNA. As an option, predictions with only poorly conserved sites are also provided. Also identified are sites with mismatches in the seed region that are compensated by conserved 3' pairing and centered sites.
DIANA Tools (http://diana.imis.athena-innovation.gr/DianaTools/index.php) is a collection of useful tools of miRNA study. This database has four powerful function:
Additionally, it also can predict signaling pathways based on multiple miRNAs and find different miRNA molecules by querying other people's data.
Besides, here, we numerate some databases related drug development and diseases.
L1000 fireworks display (L1000FWD) (http://amp.pharm.mssm.edu/L1000FWD/) is a web application that provides interactive visualization of over 16,000 drug and small-molecule induced gene expression signatures. L1000FWD enables coloring of signatures by different attributes such as cell type, time point, concentration, as well as drug attributes such as MOA and clinical phase.
MalaCards (https://www.malacards.org/) is an integrated database of human maladies and their annotations, modeled on the architecture and richness of the popular GeneCards database of human genes. It also integrates both specialized and general disease lists, including rare diseases, genetic diseases, complex disorders and more.


Comments
Leave a Comment