Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
The term "neurodegeneration" consists of the prefix "neuro-," indicating a connection to nerves or the nervous system, and "-degeneration," which, in this context, refers to a process of declining from a higher to a lower level of effective power, vitality, or essential quality. Therefore, neurodegeneration encompasses any pathological condition in which the nervous system or neuronal cells undergo a loss of function, structure, or both.
2. Four Classical Neurodegenerative Diseases
3. Common Pathogenic Mechanisms of Neurodegenerative Diseases
Neurodegeneration (ND) is a phenomenon manifested as slow and progressive functional and structural loss of neuronal cells in specific brain areas, representing the primary pathophysiological characteristic in various neurodegenerative diseases (NDD), including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), and others.
Neurodegenerative diseases involve progressive neuronal death and neurological dysfunction, resulting in escalating disability, the decline of cognitive abilities such as memory and decision-making, and motor functionalities like walking, handwriting, and swallowing, or a combination of both, depending on the specific disease. Neurodegenerative diseases typically advance gradually, often spanning several years before reaching the terminal stage. Here introduce four classical neurodegenerative diseases:
Alzheimer's disease's two major pathogenic hallmarks include the extracellular deposition of diffuse and neuritic beta-amyloid plaques and intracellular formation of hyper-phosphorylated tau neurofibrillary in the brain [1,2]. The imbalance between the production and clearance of Aβ, and the formation of tau-containing neurofibrillary tangles (NFT) disrupt the normal function of neurons and synapses, ultimately resulting in a gradual deterioration of memory, judgment, decision-making, awareness of surroundings, and the capacity for independent self-care.
Parkinson's disease has two major pathological features: the progressive loss of the dopaminergic neuromelanin-containing neurons in the substantia nigra pars compacta (SNpc) and the presence of α-synuclein-containing Lewy bodies in the cytoplasm of residual neurons [3]. These alterations culminate in tremor, rigidity, and bradykinesia/akinesia in the major muscles of the body.
ALS is characterized by progressive weakness and paralysis in striated muscles resulting from pathological degeneration of upper and lower motor neurons in the brain and spinal cord. The pathological hallmark is the accumulation of intraneuronal protein aggregates, of which the most abundant protein is the TAR DNA binding protein 43 [4, 5].
In Huntington's disease, the mHTT-containing inclusion bodies contribute to the degeneration and loss of neurons, especially in the striatum, impacting primary muscles of the body resulting in severe motor restriction and ultimately death [6].
| Neurodegenerative Diseases | Neuropathological hallmarks | Risk factors |
|---|---|---|
| Alzheimer's disease | Neuroinflammation, neuronal loss, neurofibrillary tangles, Aβ plaques | Family history, age, history of head trauma, genetics, environment factors, female gender, vascular risk factors |
| Parkinson's disease | Frontal cortex atrophy and ventricular enlargement, α-synuclein-containing Lewy bodies, and loss of dopaminergic neurons | Age, factor in the environment, male gender genetics, psychiatric symptoms ethnicity [7,8] |
| Amyotrophic lateral sclerosis | TAR DNA-binding protein 43 aggregations | Familial aggregation Physical activity, occupational and environmental exposure (for example, to pesticides, solvents, or heavy metals), genetics, head injury, and smoking |
| Huntington's disease | neuronal loss, psychiatric symptoms, striatal atrophy | Genetic mutation in HTT, inheritance |
Neurodegenerative diseases share common hallmarks, including pathological protein aggregation, synaptic and neuronal network defects, aberrant proteostasis, cytoskeletal abnormalities, altered energy and homeostasis, DNA and RNA defects, inflammation, and neuronal cell death [9].
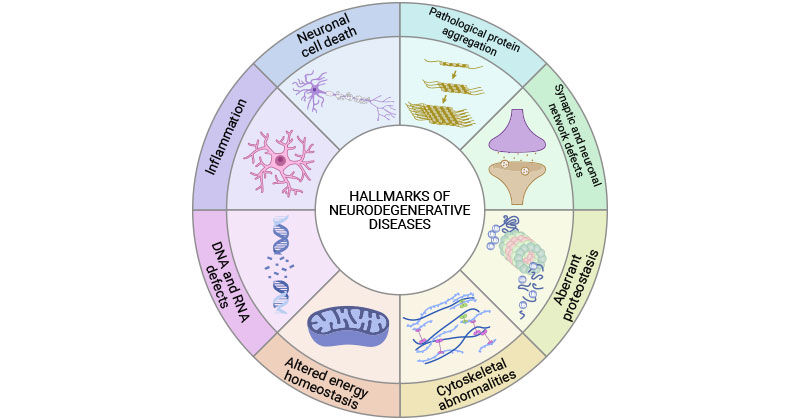
Figure 1. Common hallmarks of neurodegenerative diseases
This picture is cited from: https://www.sciencedirect.com/science/article/pii/S0092867422015756
Neurodegeneration is a complex process influenced by genetic, environmental, and endogenous factors associated with aging. However, the exact pathogenic roles and the underlying molecular mechanisms remain incompletely understood.
Common pathogenic mechanisms underlying neurodegeneration include environmental factors, metabolic stress, genetic contributions, neurovascular coupling, and neuroinflammation.
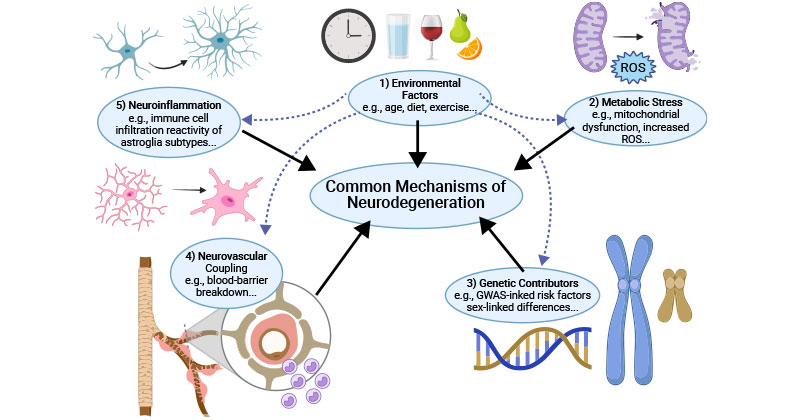
Figure 2. Common mechanisms of neurodegeneration
This picture is cited from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8935795/
Environmental factors such as age, diet, exercise, and exposure to neurotoxic substances, can trigger or exacerbate underlying neurodegenerative events, including protein misfolding and aggregation, mitochondrial dysfunction, oxidative stress, and inflammation. Age represents a primary risk factor, and tissues composed of postmitotic cells, such as neurons in the brain and retina, are especially vulnerable to the impacts of aging. Diet and exercise are increasingly recognized as crucial elements in preserving the health of the central nervous system (CNS) [10].
Metabolic stress is characterized by disrupted energy metabolism and increased oxidative stress. Neurons prone to degeneration require higher energy levels to sustain their structural and functional integrity. Hence, any disturbance in energy metabolism can generate a substantial energy demand, ultimately leading to cellular stress [11].
Mitochondria are highly and dynamically regulated to meet neuronal metabolic demands. The alterations of mitochondrial dynamics by mitochondrial dysfunction or genetic mutations will cause catastrophic influence to neurons. Mitochondrial dysfunction mainly contributes to metabolic stress due to mitochondria's vital role in oxidative energy metabolism. Increased ATP generation causes higher ROS creation.
Insufficient antioxidants, impaired scavenging enzymes, and dysfunctional mitochondria can induce oxidative stress, which impacts mtDNA, leading to an abnormality in mitochondrial quality control and electron transport chain enzymes thus causing metabolic deficiency.
In some cases, specific gene mutations are directly linked to the development of neurodegenerative diseases. For example, mutations in the APP, PSEN1, and PSEN2 genes are associated with familial Alzheimer's disease, while mutations in the HTT gene lead to Huntington's disease.
Certain genetic variations or polymorphisms may increase an individual's susceptibility to neurodegenerative diseases without directly causing the disorder. For instance, APOE4 is a common risk factor for both AD and Parkinson’s disease dementia (PDD). These genetic risk factors, often identified through genome-wide association studies, contribute to the complex interplay of genetic and environmental influences.
Many neurodegenerative diseases are polygenic, involving multiple genes with small individual effects [12]. The cumulative impact of these genetic variants, along with environmental factors, contributes to an individual's overall risk.
In the central nervous system (CNS), sex differences derive from both long- and short-term epigenetic alterations caused by gonadal hormones and their interaction with transcriptional gene products located on sex chromosomes [13,14]. Both sex hormones and sex chromosomes contribute to the CNS response to diseases and aging.
Neurovascular coupling (NVC) is the process by which cells coordinate to enhance cerebral blood flow (CBF) to meet the elevated metabolic demand of neurons during activity. NVC is essential to neurophysiologic health. NVC is mediated by multiple cell types together dubbed the neurovascular unit (NVU), including vascular cells, glial cells, and neurons, which maintain the integrity of the blood-brain barrier (BBB) and regulate the supply of the CBF.
The augmentation of CBF in response to neural activity is also proposed to contribute to the removal of waste by-products of normal function and metabolism, either through the bloodstream or perivascular mechanisms. Changes in the interplay between neural activity and CBF perturb the supply of substrates to active brain cells and hinder the elimination of potentially harmful by-products of cerebral metabolism, disrupting the cerebral microenvironment that is likely to cause brain dysfunction.
Ample studies have shown that neurovascular uncoupling is associated with the onset and progression of neurodegenerative diseases, including AD, PD, and ALS. Early pathophysiological alterations in neurodegenerative disorders, like AD, PD, and ALS, involve BBB dysfunction and reduced CBF [15,16].
Neuroinflammation not only results from neurodegeneration but also participates in this process. It significantly contributes to the pathogenesis and progression of most neurodegenerative disorders.
In AD, inflammation serves as a connection between the aggregation of Aβ and the accumulation of tau tangles [17].
In PD, alpha-synuclein aggregation, environmental toxins, or oxidative stress stimulate the activation of microglia by interacting with its Toll-like receptors [18]. The engagement of Toll-like receptors and subsequent downstream pathway activation sets off a series of events, resulting in the NF-κB activation, initiation of inflammasome formation, and elevation of cytokine levels [18]. This sequential inflammatory process results in damage to dopaminergic cells and subsequent cell death [18].
Activation of the microglial NLRP3 inflammasome has been identified as a significant contributor to neuroinflammation in ALS [19]. The NF-κB protein acts as a master regulator of inflammation in ALS. Studies have shown that TDP-43 proteinopathy induces inflammation by initiating the cytoplasmic release of mitochondrial DNA which activates the cytoplasmic DNA-sensing cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway [19,20].
Clinical manifestations of neurodegenerative diseases encompass diverse neurological symptoms such as cognitive decline, motor impairment, and behavioral changes. Neurodegenerative diseases' biomarkers include abnormal protein aggregates, neurotransmitter imbalances, and genetic variations, providing insights for diagnosis and monitoring disease progression.
| Neurodegenerative Diseases | Clinical Manifestations | Biomarkers |
|---|---|---|
| Alzheimer's disease | Memory impairment, cognitive decline, language and communication difficulties, impaired spatial and visual skills, difficulty in planning and organizing and with activities of daily living, behavior and personality changes | Aβ42, Aβ40, Tau , APOEε4, PSEN1, PSEN2 |
| Parkinson's disease | Motor symptoms including resting tremors, bradykinesia/akinesia, muscle rigidity, postural instability, masked face, speech changes; certain non-motor symptoms including cognitive impairment, mood disorders, sleep disturbances, and autonomic dysfunction | SNCA/PARK1/4, LRRK2/PARK8, PARK7/DJ-1, UCHL1/PARK5, SYNJ1/PARK20, DNAJC6/PARK19, DNAJC13/PARK21,β-Glucocerebrosidase, Aβ42, Tau, NFL, NURR1, VPS35/PARK17, EIF4G1/PARK18, BDNF, IGF-1, Let-7f-5p, miR-125a-5p, miR-151a-3p, miR-27a-3p and miR-423-5p [7] |
| Amyotrophic lateral sclerosis | Muscle weakness and atrophy, spasticity, difficulty in speaking and swallowing, impaired motor control, fasciculations, respiratory complications, emotional and cognitive changes | SOD1, FUS, C9orf72, TDP-43/TARDBP, TAF15, EWSR1, ATXN2, HnRNPA1, MATR3, TIA1/TIAR, CHCHD10, TBK1, TUBA4A, NEK1, C21orf2, CCNF, KIF5A, ANXA11, GLT8D1, SPG11, CHIT1, MiR-27a, miR-34a, miR-124, miR-142-5p, miR-155 and miR-338-3p, |
| Huntington's disease | Chorea, motor impairment, cognitive decline, behavior changes, psychiatric problems, dystonia, speech difficulties, loss of coordination, gait disturbances | HTT and mHTT |
In recent years, numerous studies about the investigation of neurodegenerative diseases' pathologies have sprung up, advancing the understanding of these complex conditions, which helps to develop effective treatments and improve the overall management and care of individuals affected by these devastating disorders.
Rising studies have demonstrated that the microbiota-gut-brain (MGB) axis exerts an important role in the pathological mechanisms of multiple neurogenerative diseases, including AD, PD, and HD. The gut microbiota mediates the physiological activities of the brain by regulating the immune system, nervous system, and microbial molecules and activating several unknown potential pathways.
Stefanie Grabrucker et al. for the first time confirmed a causal role of the gut microbiota in Alzheimer's disease, through which Alzheimer's symptoms can be transferred to a healthy young organism [21].
Van Kessel et al. found that dopamine agonist therapy in a rat model has been linked to decreased intestinal motility and small intestinal bacterial overgrowth (SIBO) and suggested that these effects are mediated by higher relative abundance of Lactobacillus and Bifidobacterium, coupled with a concurrent decline in Lachnospiraceae and Prevotellaceae [22].
Gubert et al. found that Huntington's disease mice received the transplantation of the fecal microbiota from wild-type improved cognitive outcomes, especially those females. They present evidence of sexually dimorphic and progressive gut dysfunction, along with microbial disruption, supporting the first idea of microbiota manipulation as a therapeutic intervention for Huntington's disease, especially targeting cognitive symptoms [23]. The MGB axis can impact motor, mental, and cognitive symptoms, as well as weight loss in Huntington's disease.
A CUHK research team recently discovered that the signaling pathway triggered by disturbed transcriptional factor YY1 in neurons can lead to ALS, suggesting that YY1 could serve as a potential drug target for treating this incurable disease.
In conclusion, neurodegeneration has been identified as the pivotal pathophysiological change in neurodegenerative diseases. Neurodegenerative diseases are characterized by an inexorable degeneration of a specific form of neurons. Neurodegenerative diseases are incurable and debilitating disorders, and their prevalence is rising partly due to global population aging. Therefore, understanding the complex processes involved in pathogenic mechanisms of neurodegenerative diseases is crucial for developing targeted interventions to slow or halt the progression of these conditions.
References
[1] Bayer T.A., Wirths O. Intracellular accumulation of amyloid-Beta-a predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease [J]. Front. Aging Neurosci. 2010;2:8.
[2] Pickett E.K., Herrmann A.G., et al. Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer’s disease [J]. Cell Rep. 2019;29:3592–3604.e5.
[3] Bartels A.L., Leenders K.L. Parkinson’s disease: The syndrome, the pathogenesis and pathophysiology [J]. Cortex. 2009;45:915–921.
[4] Morris J. Amyotrophic lateral sclerosis (ALS) and related motor neuron diseases: An overview. Neurodiagn [J]. J. 2015;55:180–194.
[5] Wang G., Rayner S., Chung R., Shi B., Liang X. Advances in nanotechnology-based strategies for the treatments of amyotrophic lateral sclerosis [J]. Mater. Today Bio. 2020;6:100055.
[6] Ferguson, M. W., Kennedy, C. J., et al. (2022). Current and Possible Future Therapeutic Options for Huntington’s Disease [J]. Journal of Central Nervous System Disease, 14.
[7] Emamzadeh F.N., Surguchov A. Parkinson’s disease: Biomarkers, treatment, and risk factors [J]. Front. Neurosci. 2018;12:612.
[8] Gorell J.M., Peterson E.L., Rybicki B.A., Johnson C.C. Multiple risk factors for Parkinson’s disease [J]. J. Neurol. Sci. 2004;217:169–174.
[9] David M. Wilson III, Mark R. Cookson, et al. Hallmarks of neurodegenerative diseases [J]. Volume 186, Issue 4, 16 February 2023, Pages 693-714.
[10] Businaro R, Vauzour D, et al. Therapeutic Opportunities for Food Supplements in Neurodegenerative Disease and Depression [J]. Front Nutr. 2021;8:669846.
[11] Muddapu, V. R., Parvathy Dharshini, et al. (2020). Neurodegenerative Diseases – Is Metabolic Deficiency the Root Cause [J]? Frontiers in Neuroscience, 14.
[12] Ibanez L, Farias FHG, et al. Polygenic Risk Scores in Neurodegenerative Diseases: a Review [J]. Curr Genetic Med Rep. 2019;7:22–9.
[13] Chowen JA, Garcia-Segura LM. Role of glial cells in the generation of sex differences in neurodegenerative diseases and brain aging [J]. Mech Ageing Dev. 2021;196:111473.
[14] Lentini E, Kasahara M, Arver S, Savic I. Sex differences in the human brain and the impact of sex chromosomes and sex hormones [J]. Cereb Cortex. 2013;23:2322–36.
[15] Sweeney M. D., Kisler K., Montagne A., Toga A. W., Zlokovic B. V. (2018). The role of brain vasculature in neurodegenerative disorders [J]. Nat. Neurosci. 21 1318–1331.
[16] Yu, X., Ji, C., & Shao, A. (2020). Neurovascular Unit Dysfunction and Neurodegenerative Disorders [J]. Frontiers in Neuroscience, 14.
[17] Sobue, A., Komine, O. & Yamanaka, K. Neuroinflammation in Alzheimer’s disease: microglial signature and their relevance to disease. Inflamm Regener 43, 26 (2023).
[18] Çınar, E., Tel, B. C., & Şahin, G. (2022). Neuroinflammation in Parkinson’s Disease and its Treatment Opportunities [J]. Balkan Medical Journal, 39(5), 318-333.
[19] Smethurst, P. et al. Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis [J]. Brain 143, 430–440 (2020).
[20] Mead, R. J., Shan, N., et al. (2023). Amyotrophic lateral sclerosis: A neurodegenerative disorder poised for successful therapeutic translation [J]. Nature Reviews Drug Discovery, 22(3), 185-212.
[21] Stefanie Grabrucker, Moira Marizzoni, et al. Microbiota from Alzheimer’s patients induce deficits in cognition and hippocampal neurogenesis [J]. Brain, Volume 146, Issue 12, December 2023, Pages 4916–4934.
[22] Bullock, A., & Aidy, S. E. (2022). Parkinson’s Disease Medication Alters Small Intestinal Motility and Microbiota Composition in Healthy Rats [J]. MSystems, 7(1).
[23] Gubert, C., Choo, J. M., et al. (2022). Faecal microbiota transplant ameliorates gut dysbiosis and cognitive deficits in Huntington’s disease mice [J]. Brain Communications, 4(4).


Comments
Leave a Comment