Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Literature Title: SKAP2 restores sperm motility and morphology through modulating mitochondrial organization and cytoskeletal remodeling
Journal: Signal Transduct Target Ther
Impact Factor: 52.7
Client Institutions: Zhejiang University, Fudan University
Male infertility has become a global health issue, with asthenoteratozoospermia being a common cause. Although some cases are associated with genetic mutations, the molecular mechanisms remain unclear, and targeted treatment strategies are lacking. On December 24, 2025, a research team from Zhejiang University and Fudan University published a research paper titled "Targeting SKAP2 restores sperm motility and morphology through modulating mitochondrial organization and cytoskeletal remodeling" in the journal Signal Transduction and Targeted Therapy. Through systematic screening and functional validation, the study first revealed the core regulatory role of the HNRNPR gene and its downstream target SKAP2 in spermatogenesis. It innovatively proposed a protein delivery strategy based on extracellular vesicles (mEVs), providing new insights for the treatment of male infertility.
1. First revelation of the HNRNPR-SKAP2 axis's critical role in spermatogenesis, elucidating a new mechanism of m6A-dependent splicing.
2. Breakthrough therapeutic strategy: Utilizing natural extracellular vesicles to deliver SKAP2 protein, enabling non-invasive repair of sperm functional abnormalities caused by genetic defects.
3. Clinical translational potential: Validating the therapeutic effect of mEVs-SKAP2 in human sperm, providing a precise intervention target for asthenoteratozoospermia.
The research team recruited 572 asthenoteratozoospermia patients. Whole-exome sequencing (WES) was performed on three infertile men from two families (one consanguineous and one non-consanguineous). Homozygous missense mutations (c.1540 G > A) in the HNRNPR gene were identified in two brothers from the consanguineous family (Fig. 1b, c). Compound heterozygous missense mutations (c.1280 A > C, c.1369 G > A) in the same gene were found in the proband from the non-consanguineous family (Fig. 1d, e).
Sanger sequencing confirmed the co-segregation of these mutations with the disease phenotype within the families (i.e., patients carried the mutations, parents were heterozygous carriers, and healthy relatives did not carry them). Protein domain localization of the mutation sites (Fig. 1f) identified that the mutations were located in or near the RGG domain of the hnRNPR protein, which is crucial for RNA binding function. Sperm phenotype analysis (Fig. 1g-j) using CASA showed that, although sperm concentration was normal, both progressive motility and the proportion of morphologically normal sperm were significantly reduced in mutation carriers, consistent with the clinical diagnosis of asthenoteratozoospermia (Fig. 1g, h). Morphological staining and TEM revealed widespread morphological abnormalities in sperm, including ultrastructural defects such as acrosomal detachment and disorganized neck structure (Fig. 1i, j). This study demonstrated that mutations in the HNRNPR gene are a novel genetic cause of asthenoteratozoospermia and male infertility.
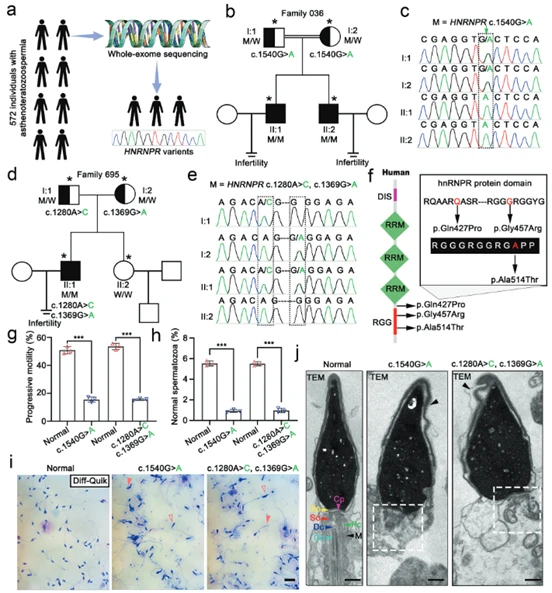
(Fig. 1: HNRNPR gene mutations cause asthenoteratozoospermia and male infertility)
Using gene editing technology, a gene knock-in (KI) mouse model carrying the Hnrnpr pathogenic mutation was constructed. Results showed no significant differences in body size or the ratio of testis weight to body weight between KI and wild-type (WT) mice (Fig. 2b and supplementary figures), indicating the mutation did not affect overall growth or testis size. Fertility tests revealed that male mice with the Hnrnpr mutation failed to produce any offspring after co-housing with female mice for four months, exhibiting complete sterility (Fig. 2b). PAS staining results showed no significant difference in seminiferous tubule structure or total sperm count between KI and WT mice (Fig. 2c-f), indicating the Hnrnpr mutation does not affect sperm production quantity. CASA analysis showed that, despite normal sperm counts, both total motility and progressive motility of KI mouse sperm were significantly lower than those of WT mice (Fig. 2g, h), recapitulating the core features of human asthenoteratozoospermia. These results demonstrate that Hnrnpr mutation also leads to male infertility in the mouse model. This infertility stems not from impaired spermatogenesis or reduced sperm numbers, but from severe defects in sperm motility function, successfully reproducing the pathological phenotype caused by HNRNPR mutation in humans in vivo.
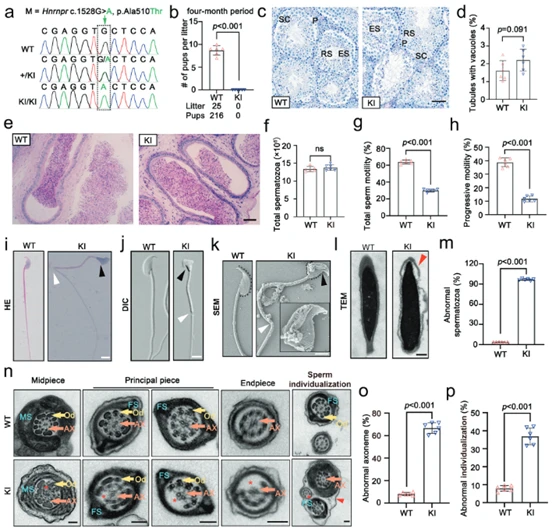
(Fig. 2: Hnrnpr gene mutation leads to asthenoteratozoospermia and male infertility in mice)
PAS and PNA staining on testicular sections from wild-type (WT) and mutant (KI) mice first revealed obvious acrosomal defects at step 9 of spermatid development (Fig. 3a), indicating the Hnrnpr mutation causes impact at an early stage of sperm formation. Using transmission electron microscopy (TEM) for high-resolution imaging of spermatids at different developmental stages, results showed that WT mice had normal acrosome formation, migration, and nuclear elongation, whereas KI mice exhibited significant acrosomal malformation, with developing acrosomes mispositioned (ectopic) and failing to correctly attach to the nuclear surface (Fig. 3b). By TEM observation of the manchette (a temporary microtubular structure crucial for shaping the sperm head and flagellum) and α-tubulin immunofluorescence for localization and length quantification, it was found that the manchette in KI mice was abnormally positioned, excessively elongated, and structurally asymmetric, with its microtubule length significantly longer than in controls (Fig. 3c-e). Cellular and ultrastructural analyses demonstrate that hnRNPR protein is crucial for proper acrosome attachment, normal manchette organization, and successful nuclear elongation during spermatogenesis. Hnrnpr mutation disrupts these key morphogenetic steps, ultimately leading to sperm malformation.
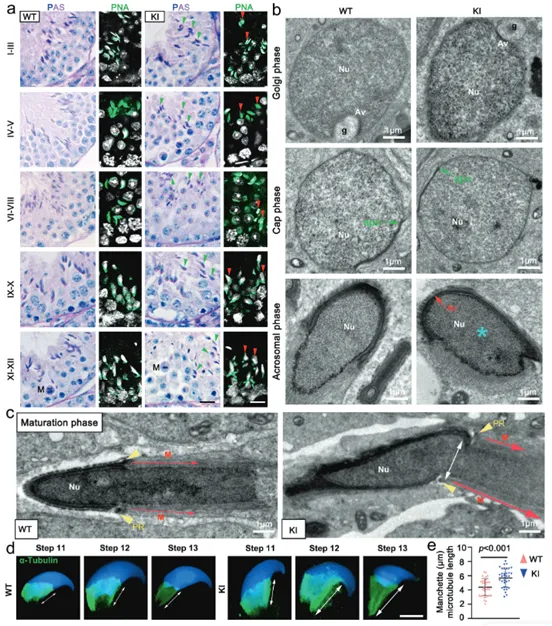
(Fig. 3: Abnormalities in acrosome, nucleus, and manchette assembly in gene knock-in mouse sperm)
Single-cell RNA sequencing on round spermatids from wild-type (WT) and mutant (KI) mice identified 498 differentially expressed genes (DEGs) in KI mice, with 261 downregulated and 237 upregulated (Fig. 4a). Gene Ontology (GO) analysis showed these DEGs were significantly enriched in biological pathways related to cytoskeleton organization and sperm motility (Fig. 4b). Among the downregulated genes, Skap2 expression was significantly reduced in KI mice (Fig. 4c). RT-qPCR experiments confirmed that Skap2 mRNA levels were indeed significantly decreased in round spermatids of mutant mice (Fig. 4d). Comparative proteomic analysis of human and mouse (KI/WT) testes and mature sperm identified 63 common differentially expressed proteins across different species and tissue samples (Fig. 4e). These overlapping proteins were also significantly enriched in pathways like sperm cytoskeleton organization and F-ACTIN assembly (Fig. 4f), highly consistent with transcriptomic results. Integrating transcriptomic and proteomic data, SKAP2 was identified as a key molecule altered at both RNA and protein levels (Fig. 4g). Among the differentially expressed proteins, up to 71.08% were related to the sperm cytoskeleton, of which 77.15% were associated with acrosome formation and function (Fig. 4h). Heatmaps further displayed expression changes in various cytoskeletal and F-ACTIN-related proteins (Fig. 4i, j). Western blot analysis on isolated round spermatids, consistent with omics data, showed SKAP2 and F-ACTIN protein levels were significantly reduced in KI mice (Fig. 4k, l). Multi-omics analysis strongly demonstrated that Hnrnpr mutation leads to downregulation of SKAP2 and F-ACTIN expression at both transcriptional and translational levels. These molecular-level changes directly explain defects in sperm cytoskeletal remodeling, constituting the key molecular mechanism leading to abnormal sperm morphology and reduced motility.
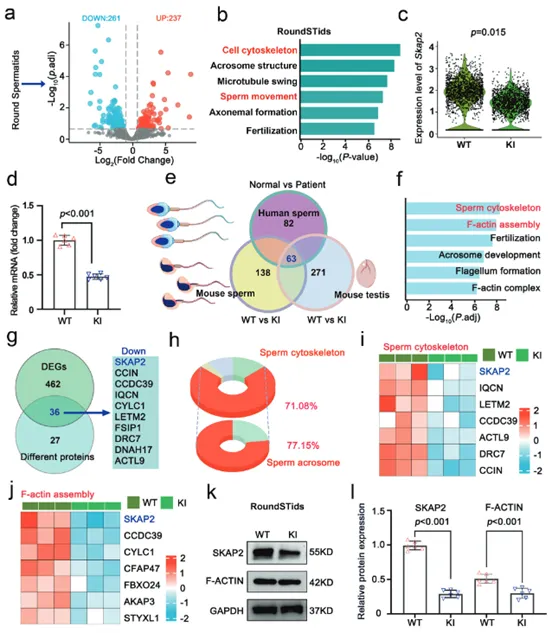
(Fig. 4: Abnormal RNA and protein profiles in gene knock-in mice)
RNA immunoprecipitation sequencing (RIP-seq) was used to identify RNA targets directly bound by hnRNPR protein in round spermatids. The sequence features and distribution positions of RNA regions bound by hnRNPR highly overlapped with known m6A modification sites (Fig. 5a-e), suggesting hnRNPR may function as an m6A "reader". Integrating RIP-seq, m6A-seq, and differential splicing analysis data, Skap2 was identified as a key gene simultaneously meeting the three criteria: "bound by hnRNPR", "m6A-modified", and "aberrantly spliced in mutant mice" (Fig. 5f). Further analysis revealed that the Skap2 gene in mutant mice exhibited skipping of exon 2, i.e., a splicing error. A minigene reporter system containing the Skap2 gene fragment was constructed, and its m6A modification sites were specifically disrupted. Observing splicing changes in cells showed that disrupting the m6A sites also caused skipping of Skap2 exon 2, successfully recapitulating the splicing defect of mutant mice in vitro (Fig. 5g-k). A series of evidence proves that hnRNPR ensures correct splicing of Skap2 by recognizing m6A modification sites on its mRNA. The Hnrnpr mutation disrupts this precise regulatory process, leading to aberrant Skap2 splicing, which is a key downstream pathogenic event.
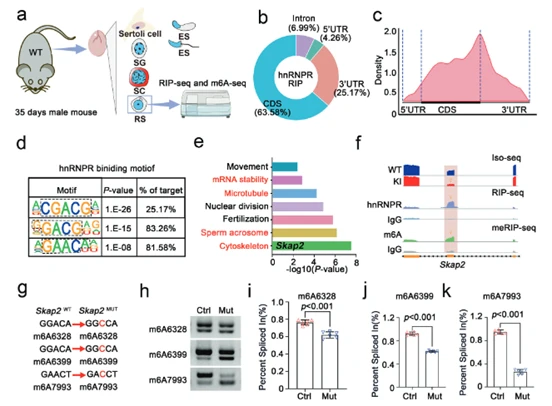
(Fig. 5: hnRNPR protein regulates Skap2 transcript alternative splicing via m6A mediation)
Since systemic knockout of Skap2 leads to embryonic lethality in mice, researchers constructed a conditional gene knockout mouse model with Skap2 specifically deleted in male germ cells. RT-qPCR, Western blot, and immunofluorescence (IF) experiments successfully confirmed effective knockout of SKAP2 protein in germ cells of mutant mice (Fig. 6b-e). Fertility tests by co-housing knockout (Skap2 D-cKO) male mice with wild-type females showed that Skap2 D-cKO male mice were completely sterile, confirming SKAP2's essential role in male fertility (Fig. 6n). Sperm morphological analysis using TEM revealed that most sperm flagella lacked the crucial "9+2" axonemal microtubule structure, the fundamental structural defect preventing normal movement (Fig. 6f-i). Additionally, Fig. 6l, m shows abnormal development of the manchette structure, crucial for sperm head shaping. Computer-assisted sperm analysis (CASA) was used to quantitatively assess sperm motility parameters. Compared to wild-type mice, sperm from Skap2 D-cKO mice showed significantly decreased total motility and progressive motility, exhibiting typical asthenozoospermia features (Fig. 6j, k). These results demonstrate that specific knockout of the Skap2 gene in male germ cells is sufficient to perfectly recapitulate all key phenotypes caused by Hnrnpr mutation (including lack of sperm tail structure, low motility, and male sterility). This ultimately confirms that SKAP2 is the core executor in the hnRNPR-SKAP2 signaling axis directly responsible for sperm abnormalities.
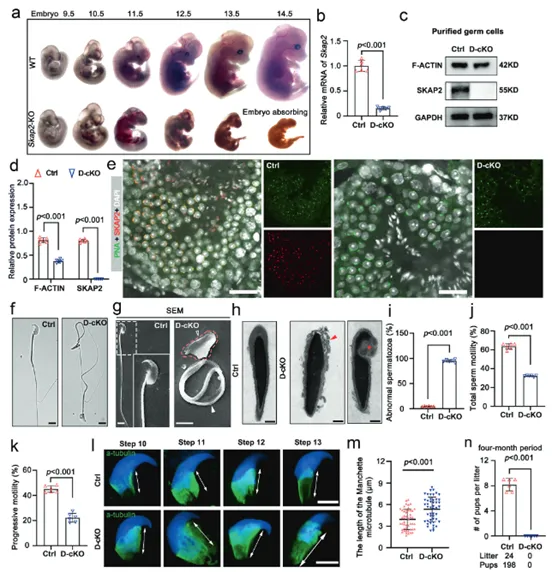
(Fig. 6: SKAP2 deficiency interferes with spermatogenesis and causes sperm malformation)
Extracellular vesicles loaded with SKAP2 protein (mEVs-SKAP2) were microinjected into the efferent ducts of Hnrnpr mutant (KI) mouse testes. CASA was then used to evaluate therapeutic effects. Results showed that compared to untreated or empty-vesicle control groups, mice treated with mEVs-SKAP2 exhibited significantly improved total sperm motility and progressive motility, along with a significantly reduced proportion of morphologically abnormal sperm (Fig. 7a-d). TEM observation of ultrastructural changes in treated sperm revealed that mEVs-SKAP2 treatment significantly reduced morphological defects like sperm tail bending and acrosomal detachment, but did not repair the core "9+2" microtubule structure of the axoneme (Fig. 7f-j), indicating the therapy primarily improves secondary structural defects. Immunofluorescence staining was used to detect and quantify the assembly of the key cytoskeletal protein F-ACTIN in sperm cells. mEVs-SKAP2 treatment significantly restored normal assembly and localization of F-ACTIN (Fig. 7k). This explains, at the molecular level, the improvement in sperm morphology and motility, i.e., SKAP2 functions by repairing the cytoskeleton. Results prove that mEVs-SKAP2 is an effective in vivo therapeutic strategy. It can significantly improve sperm motility and partially ameliorate morphological defects by repairing cytoskeletal structure (F-ACTIN), providing a new potential solution for treating male infertility caused by specific gene mutations.
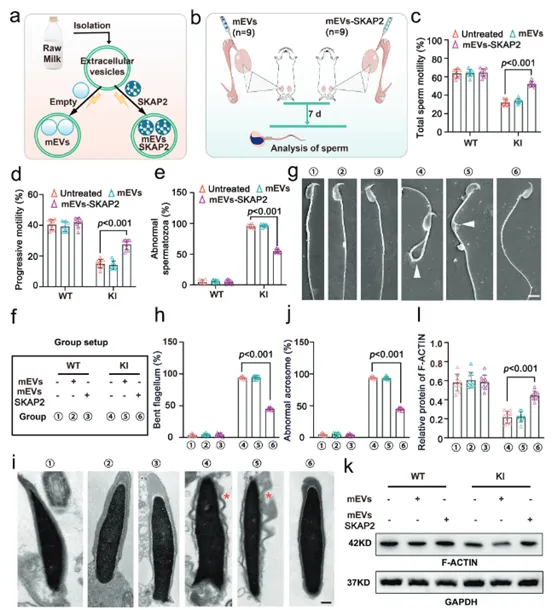
(Fig. 7: Extracellular vesicle-SKAP2 restores sperm motility and morphology in Hnrnpr mutant mice)
Using freeze-fracture combined with scanning electron microscopy, the mitochondrial ultrastructure in the midpiece of mouse sperm tails was observed at high resolution. In untreated Hnrnpr mutant (KI) mice, the mitochondrial sheath in the sperm tail was disorganized and loosely arranged, failing to form a normal compact structure. After mEVs-SKAP2 treatment, sperm mitochondrial structure was significantly improved (Fig. 8a-d). This study reveals the therapeutic mechanism of mEVs-SKAP2 at the mitochondrial level. The therapy effectively repairs the organizational structure of the sperm mitochondrial sheath, making its arrangement tighter and more orderly. This structural optimization helps improve ATP production and supply efficiency, thereby providing a solid energy foundation for the restoration of sperm motility.
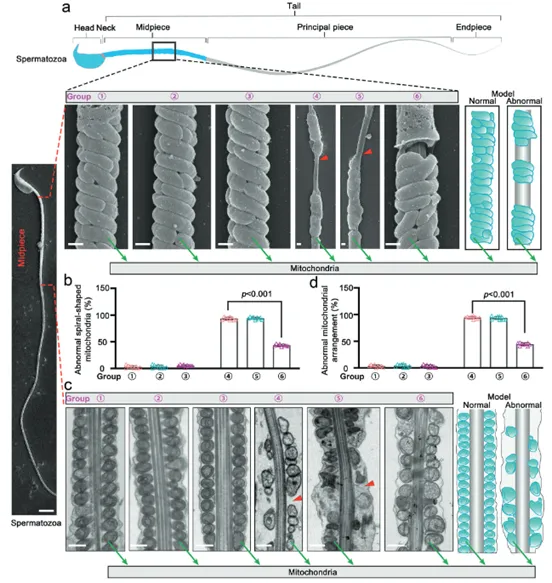
(Fig. 8: mEVs-SKAP2 restores mitochondrial structure in mouse sperm)
Extracellular vesicles loaded with SKAP2 (mEVs-SKAP2) were co-cultured with human sperm from different sources (healthy individuals, HNRNPR mutation patients, and other asthenoteratozoospermia patients). CASA was then used for detection. Results showed that compared to the untreated group, mEVs-SKAP2 treatment significantly enhanced total motility and progressive motility of human sperm in all groups, indicating the therapy is also effective in vitro (Fig. 9b, c, e). Morphological analysis of the treated human sperm showed that mEVs-SKAP2 had limited effect on overall sperm morphology but significantly reduced the proportion of sperm with tail bending (Fig. 9d, e). Using TEM to observe human sperm tail structure after in vitro treatment yielded results consistent with in vivo treatment: while mEVs-SKAP2 partially alleviated tail bending, the core "9+2" axonemal microtubule structure was not repaired (Fig. 9e). The same results were observed in in vitro experiments using the mouse model. Immunofluorescence staining to detect polymerization of the key cytoskeletal protein F-ACTIN in in vitro treated human and mouse sperm found that mEVs-SKAP2 treatment could significantly promote F-ACTIN protein polymerization, providing direct molecular mechanism evidence for the restoration of sperm motility in vitro (Fig. 9f, g). Research results indicate that the mEVs-SKAP2 therapy is effective for both human and mouse sperm in vitro. It significantly improves sperm motility and partially ameliorates morphological defects (e.g., tail bending) by remodeling the cytoskeleton (promoting F-ACTIN polymerization), but cannot repair the core axonemal structure of the sperm tail. This demonstrates the broad application prospects of this therapy in clinical assisted reproductive technologies.
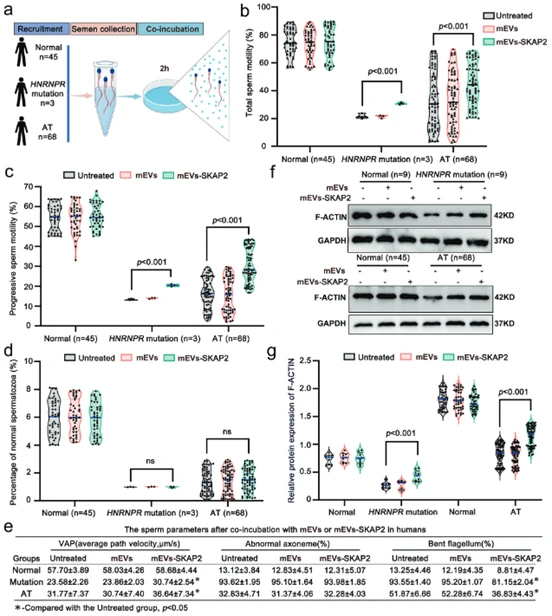
(Fig. 9: mEVs-SKAP2 alleviates asthenoteratozoospermia in human sperm)
This study reveals that the HNRNPR-mediated, m6A-dependent RNA splicing regulation of SKAP2 is a key pathogenic mechanism causing asthenoteratozoospermia. Based on this, a novel extracellular vesicle-based SKAP2 protein delivery therapy was developed, providing a comprehensive new strategy from molecular mechanism to targeted therapy for male infertility.
In this study, researchers used CUSABIO's TUBA1A Monoclonal Antibody (Product Code: CSB-MA754656A0m) to detect α-Tubulin expression in immunofluorescence experiments.

CUSABIO is committed to providing biological reagents that satisfy global researchers, supporting innovation and translation in life science research.
References
Targeting SKAP2 restores sperm motility and morphology through modulating mitochondrial organization and cytoskeletal remodeling. Signal Transduct Target Ther, 2025 Dec 24.


Comments
Leave a Comment