Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
| Code | CSB-PA009125LA01HU |
| Size | US$166 |
| Order now | |
| Image |
|
| Have Questions? | Leave a Message or Start an on-line Chat |
The GAA Antibody (Product code: CSB-PA009125LA01HU) is Non-conjugated. For GAA Antibody with conjugates, please check the following table.
| Application | Recommended Dilution |
|---|---|
| WB | 1:1000-1:5000 |
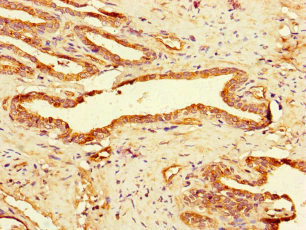
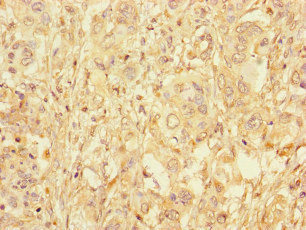
| IHC | 1:20-1:200 |
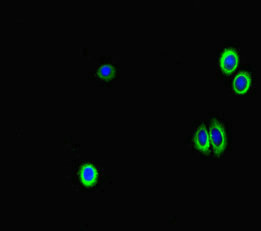
| IF | 1:50-1:200 |
The antibody GAA is generated in rabbits by immunization with a peptide spanning amino acids 601-952 of the recombinant human lysosomal alpha-glucosidase protein. This antibody exists as an unconjugated IgG and can bind to both human and mouse samples. Lysosomal alpha-glucosidase, the target protein of this GAA antibody, is an enzyme that plays a critical role in the lysosomal glycogen breakdown. Besides its function in glycogen metabolism, lysosomal alpha-glucosidase is also involved in autophagy regulation.
The GAA antibody is highly pure, with a protein G purification level of over 95%. It has been extensively tested and validated for use in ELISA, WB, IHC, and IF assays, enabling the detection and localization of lysosomal alpha-glucosidase.
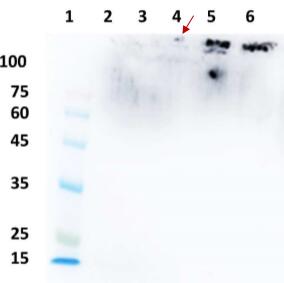
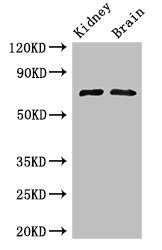
Applications : Immunoblot analysis
Review: Supernatants were collected and loaded onto SDS-PAGE gels, and proteins transferred to nitrocellulose membranes, immunoblotted using glucosidase antibody.
By Anonymous

