Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
When talking to lysosomal enzymes, we have to first describe lysosomes. Lysosomes were first isolated from rat liver cells using cell fractionation techniques by Christian de Duve and his team in 1955. Lysosomes are membrane-bound organelles present in most eukaryotic cells. Lysosomes function as the digesters of a eukaryotic cell due to the presence of diverse hydrolytic enzymes. Their luminal pH is about 4.5, providing an acidic environment for the optimal activity of hydrolytic enzymes.
This text will focus on the definition, biosynthesis and transport, classification, function, and other aspects of lysosomal enzymes. The comparison of the lysosomal enzymes with other two enzymes such as lysozyme and peroxidase will also be described. Finally, lysosomal storage diseases and related therapies will be introduced.
1. What Are Lysosomal Enzymes?
2. Biosynthesis and Transport of Lysosomal Enzymes
3. What Are the Types of Lysosomal Enzymes?
4. What Are functions of Lysosomal Enzymes?
Lysosomal enzymes are a vast array of hydrolases found in the lysosomes within most eukaryotic cells.
Most lysosomal enzymes are first synthesized as proenzymes in ribosomes of the rough endoplasmic reticulum (RER). The proenzymes contain an N-terminal sequence of 20-25 amino acids that directs their translocation into the lumen of the ER [1]. Then, the signal peptide is removed by signal peptidase. Concomitantly, proenzymes undergo N-linked glycosylation by transferring a preformed oligosaccharide from dolochol-P-P-oligosaccharide (DoI). These proteins traverse the ER-Golgi intermediate compartment by vesicular transport. When arriving in the cis-Golgi apparatus, the mannose on the lysosomal enzymes are phosphorylated by the sequential action of two enzymes: GlcNac-1-phosphotransferase and N-acetylglucosamine-1-phosphodiester α N-acetylglucosaminidase, forming the mannose 6-phosphate (M6P), the recognition signal for lysosomal enzyme sorting. The M6P tag is recognized in the trans-Golgi network (TGN) by M-6-P receptors (M6PRs) that target the lysosomal enzymes to the endo-lysosomal pathway [2-5]. In this way, lysosomal enzymes are packed into clathrin-coated vesicles that bud from the trans-Golgi network in the form of primary enzymes. The vesicles subsequently deliver their contents to a late endosome, where the primary enzymes dissociate from the M6PRs at the acidic internal pH. The enzymes are thus released into the lumen of the endosome, while M6PRs remain in the membrane and are eventually recycled to the Golgi. The acidic environment makes these enzymes biologically active.
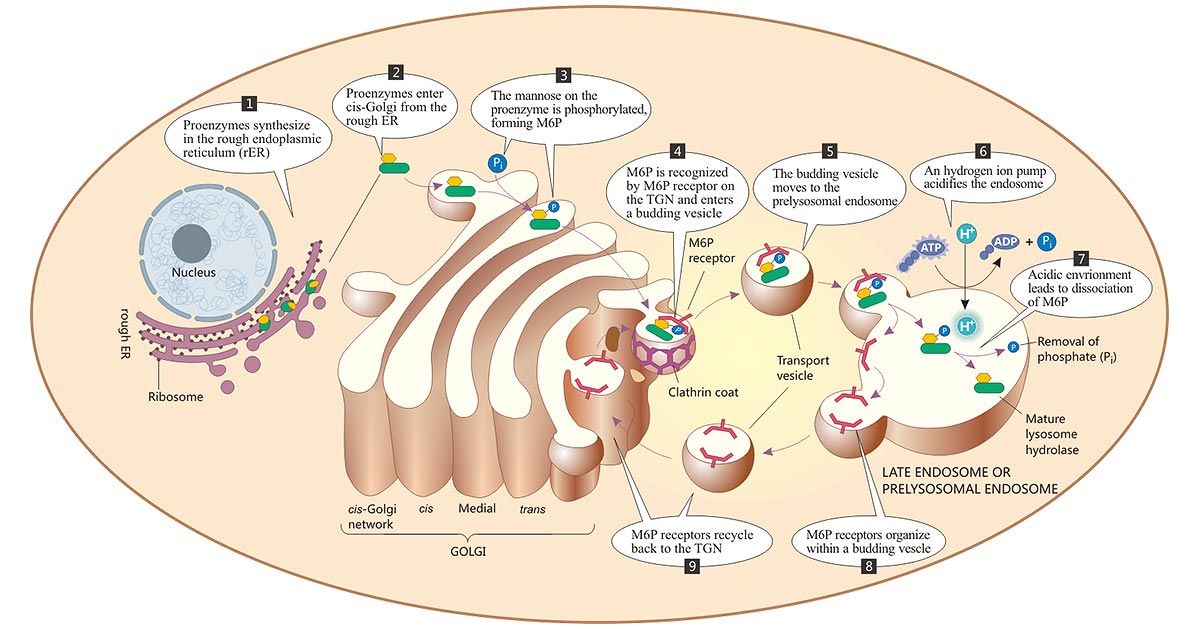
Figure: The biosynthesis and transport of lysosomal enzymes
The picture is sourced from https://doctorlib.info/physiology/medical/6.html
Lysosomes contain about 60 types of hydrolytic enzymes, including glycosidases, proteases, nucleases, lipases, phosphatases, proteases, and sulfatases. All are acid hydrolases. They require an acidic environment for optimal activity, which the lysosome supplies by maintaining a pH of around 5.0 in its core.
Table:The classification of lysosomal enzymes
Lysosomes and lysosomal enzymes play a central role in diverse cellular processes, including cellular maturation, recycling, signaling, defense, and cell death. Lysosomes enzymes are responsible for the degradation of most biological molecules, including proteins, carbohydrates, nucleic acid, lipids, and other building blocks of protoplasm. They also assist in destroying cell debris taken up by the cell during the process of endocytosis.
Lysosomes contain many enzymes with strong hydrolytic capacity but do not occur autolysis neither nor normally damage the cell itself. Because the lysosomal membrane is endowed with diverse acidic, highly glycosylated membrane integrins, whose oligosaccharide chain extends to the inner surface of the lysosomes, protecting lysosomes from hydrolysis by the hydrolytic enzymes. The lysosomal membrane has a high content of cholesterol, promoting the stability of the membrane structure. Furthermore, the requirement for acidic pH by these lysosomal enzymes provides double protection against uncontrolled digestion of the cytosol's contents. Even when the lysosome membrane is destroyed, the released acid hydrolase is inactive at neutral cytoplasmic pH.
Lysosomal enzymes are acidic hydrolases found in the lysosomes and require acidic pH (ranging from 4.5-5.0) for their optimal activity. They are responsible for the degradation of biological polymers such as proteins, lipids, polysaccharides, and nucleic acids. The enzymes destined in the lysosomes are labeled with M6P. They are synthesized in the ribosomes in the rough endoplasmic reticulum and transported through the Golgi apparatus to the trans-Golgi network. The transport vesicles deliver these proteins to late endosomes bud from the trans-Golgi network through the interactions between M6P and M6P receptors. In the acidic environment, the phosphate is cleaved, and the enzymes are released and become mature forms.
Lysozymes, also called muramidases or N-acetylmuramylhydrolases, are hydrolytic glycosidases secreted by submucosal glands, neutrophils, and macrophages. In mammals, abundant lysozymes are found in the blood and liver, in body secretions such as tears, saliva, urine, and milk, and in professional phagocytes, including macrophages, neutrophils, and dendritic cells [6] [7]. Lysozymes exert antibacterial, anti-inflammatory, antiviral, and other effects, and are mainly used for innate immunity and host defense. They act as an antimicrobial agent by hydrolyzing the β-1,4 glycosidic bonds between N-acetylmuramic acid (NAM) and N-acetylglucosamine (NAG) of cell wall peptidoglycan (PG) in Gram-positive and Gram-negative bacteria, which leads to the loss of cellular membrane integrity and cell death [8] [9]. Additionally, hydrolytic products can boost IgA secretion, macrophage activation, and bacterial pathogen clearance [10]. Against most bacteria, lysozyme acts synergistically with other antimicrobial polypeptides. In addition to hydrolytic muramidase activity, most lysozymes also possess chitinase activity, probably as a consequence of the similarity between peptidoglycan and chitin.
Peroxidases are oxidative enzymes located in peroxisomes within all eukaryotes. They are responsible for oxidizing organic compounds and breaking down metabolic hydrogen peroxides. Free ribosomes produce the proteins destined for peroxisomes, and these proteins are obtained from the cytosol. These proteins are labeled with peroxisomal targeting signal (PTS) in the cytosol. The target proteins' C-terminus is tagged with PTS1 and their N-terminus with PTS2, and then the cargo proteins Pex5 and Pex7 respectively carry them into peroxisomes.
Defects in genes coding for lysosomal enzymes may cause lower or absent catalytic activity, less stable protein folding, ER retention, and aberrations in lysosomal intracellular trafficking and signaling, all of which lead to the accumulation of the undigested substrates in the cytosol, eventually resulting in lysosomal storage disorders in humans such as Gaucher's disease, cardiovascular diseases, neurodegenerative disorders and several cancers [11].
Lysosomal storage disease not only affects the normal function of a certain organ of the body but also often affects the coordination of metabolic activities of the entire body, leading to a variety of diseases.
Gaucher's disease results from a mutation in the homozygous glucocerebrosidase (GBA) gene encoding the glucosylce¬ramidase (GCase). The GCase is required for the breakdown of glucosylceramide (GlcCer) into ceramide and glucose. Farber disease is caused by deficiency of acid ceramidase and storage of undegraded ceramides, particularly, ceramides containing 2-hydroxy fatty acids, in lysosomes. Krabbe disease is a demyelin¬ating LSD caused by a deficiency in galac¬tosylceramidase with the accumulation of galactosylceramide and galactosylsphingosine. Glycogen storage disease (GSD) is caused by the lack of an acid II-glucosidase in the lysosomes of liver and muscle cells. Normally, this enzyme decomposes glycogen. When this enzyme is lacking, the excess glycogen taken in by the lysosome cannot be degraded, which causes a large amount of glycogen to accumulate in the secondary lysosome, leading to lysosome swelling, eventually causing the lysosome to rupture and other enzymes to leak out. This would result in serious damage.
Lysosomal overload may cause lysosomal enzyme instability, leading to their leakage into the cytosol. Lysosomal enzymes like RNase and DNase have genotoxic potential because they may interact with the nucleic acid. Their abnormal leakage may induce cell apoptosis or oncosis.
Christian de Duve proposed the idea that LSDs could be treated by replacing the defective enzymes with their normal counterparts in 1964 [12]. Subsequent experiments showed that the exogenous enzyme obtained access to and degraded the substrates stored in the lysosomes when the appropriate active enzyme was introduced to the media of enzyme-deficient cultured fibroblasts from patients with certain LSDs [13-15]. Later, it was found that lysosomal enzymes were targeted to lysosomes through the M6P receptor-mediated pathway. The discovery of M6P receptor-mediated cellular uptake and delivery of normal enzymes administered intravenously to lysosomes further provided a theoretical basis for enzyme replacement therapy (ERT) in the treatment of non-neural LSDs. Available therapies based on enzyme replacement may lead to stabilization and even partial recovery in some patients.
References
[1] Thomas Braulke and Juan S.Bonifacino. Sorting of lysosomal proteins [J]. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research Volume 1793, Issue 4, April 2009, Pages 605-614.
[2] P. Ghosh, N.M. Dahms, S. Kornfeld. Mannose 6-phosphate receptors: new twists in the tale [J]. Nat. Rev. Mol. Cell Biol., 4 (2003), pp. 202-212.
[3] Hasilik A, Wrocklage C, Schröder B. Intracellular trafficking of lysosomal proteins and lysosomes [J]. Int. J. Clin. Pharmacol. Ther. 2009, 47(Suppl. 1): S18–33.
[4] Kornfeld S. Lysosomal enzyme targeting [J]. Biochem. Soc. Trans. 1990, 18: 367–74.
[5] von Figura K. Molecular recognition and targeting of lysosomal proteins [J]. Curr. Opin. Cell Biol. 1991, 3: 642–46.
[6] Ragland SA, Criss AK. From bacterial killing to immune modulation: Recent insights into the functions of lysozyme [J]. PLoS Pathog. 2017;13(9):e1006512.
[7] Callewaert L and Michiels CW. Lysozymes in the animal kingdom [J]. J Biosci. 2010 Mar; 35(1):127-60.
[8] Ellison III RT, Giehl TJ. Killing of gram-negative bacteria by lactoferrin and lysozyme [J]. J Clin Invest. 1991;88:1080–91.
[9] Melani Solomon and Silvia Muro. Lysosomal Enzyme Replacement Therapies: Historical Development, Clinical Outcomes, and Future Perspectives [J]. Adv Drug Deliv Rev. 2017 Sep 1; 118: 109–134.
[10] Clarke TB, Davis KM, et al. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity [J]. Nat Med. 2010;16:228–31.
[11] Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nature reviews Molecular cell biology [J]. 2004;5:554–565.
[12] de Duve C. From cytases to lysosomes [J]. Fed. Proc. 1964, 23:1045–49.
[13] Cantz M, Kresse H. Sandhoff disease: defective glycosaminoglycan catabolism in cultured fibroblasts and its correction by β-N-acetylhexosaminidase [J]. Eur. J. Biochem. 1974, 47: 581–90.
[14] O'Brien JS, Miller AL, et al. Sanfilippo disease type B: enzyme replacement and metabolic correction in cultured fibroblasts [J]. Science 1973, 181: 753–55.
[15] Porter MT, Fluharty AL, Kihara H. Correction of abnormal cerebroside sulfate metabolism in cultured metachromatic leukodystrophy fibroblasts [J]. Science 1971, 172: 1263–65.
Comments
Leave a Comment


Thank you for your article. I believe that Lysosomal dysfunction is much more common as we age and remains undocumented, More research is needed in this area.