Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Anaplastic lymphoma kinase (ALK) is a transmembrane receptor tyrosine kinase of the insulin receptor superfamily. It is highly homologous with leukocyte tyrosine kinase [1].
ALK plays a major role in the development of the central and peripheral nervous systems. ALK promotes apoptosis in the absence of ligand binding and inhibits apoptosis in the presence of ligand binding (or ALK fusion protein). ALK abnormality is an important factor for the occurrence and development of tumors. Its abnormality mainly includes gene fusion, gene mutation, gene amplification and protein expression increase, among which gene fusion is the most common. ALK abnormality occurs in lymphoma, neuroblastoma, non-small cell lung cancer (NSCLC) and other tumors, and is the oncogenic driver gene of tumors.
2. Abnormalities in The ALK Gene
3. ALK Gene Rearrangement and Tumor
4. Gene Copy Number Variation and Tumor
5. The Detection Method of ALK Fusion Gene
In 1994, ALK was first discovered in the form of the NPM1NPM1-ALK fusion gene in large-cell lymphoma (ALCL) [2]. Therefore, it was named as anaplastic lymphoma kinase. ALK is highly conserved in all species. The gene is located on human chromosome 2p23 and is approximately 728 kb in length and contains 29 exons. The cDNA is normally 6226 bp, encoding a protein of 177 kDa, and the mature ALK protein after translation modification is about 200-220 kDa.
ALK is a single chain transmembrane protein containing 1620 amino acids, consisting of three parts: extracellular region (aa 1~1030), transmembrane region (aa 1030~1058), and intracytoplasmic kinase catalytic region (aa 1058~1620) [3].
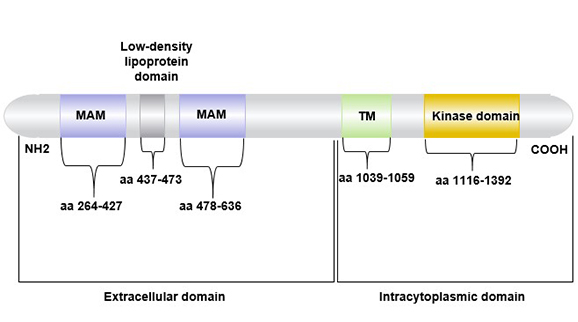
Figure 1 The structure of ALK(anaplastic lymphoma receptor tyrosine kinase)
ALK is expressed in the adult brain, which has an important effect on the development and function of the nervous system. In addition, ALK is also expressed in the small intestine, testis, prostate and colon, but it is not expressed in normal lymphoid tissues, lungs and other tissues.
ALK has abnormal changes in a variety of tumors, including anaplastic large cell lymphoma (ALCL), rhabdomyosarcoma, inflammatory myofibroblastoma, neuroblastoma and non-small cell lung cancer (NSCLC). Gene fusion is the main form of ALK abnormality. In addition, increased gene amplification/copy number and point mutations in the kinase domain also enhanced the abnormal activity of ALK kinase. Up to now, 21 different genes have been found to translocate with ALK, and different ALK fusion proteins may cause the activation of different signaling pathways.
ALK activates multiple intracellular signaling pathways, and the known types of pathways activated by ALK include: Phospholipase C gamma, JAK-STAT3, phosphatidyl inositol - 3 - kinase (PI3K), mammalian target of rapamycin(mTOR), and mitogen-activated protein kinase (MAPK). Activation of these signaling pathways is involved in regulating cell growth, transformation and anti-apoptosis [4].
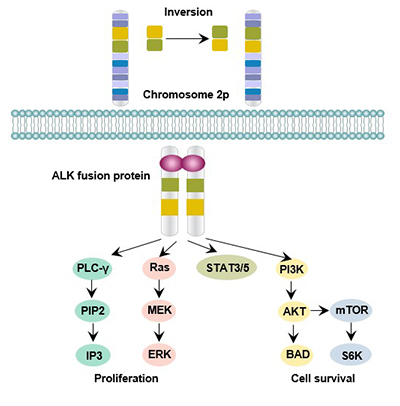
Figure 2 ALK-mediated signaling pathway
ALK gene recombination can be used as a tumor driver [5]. ALK induces the formation of malignant tumor phenotypes by promoting the activation of downstream signaling pathways and the proliferation of tumor cells. ALK gene recombination may be caused by double-stranded DNA break and abnormal DNA terminal connection [6]. In general, different ALK fusion genes show differences in tumor transformation and tumorigenesis potential [7].
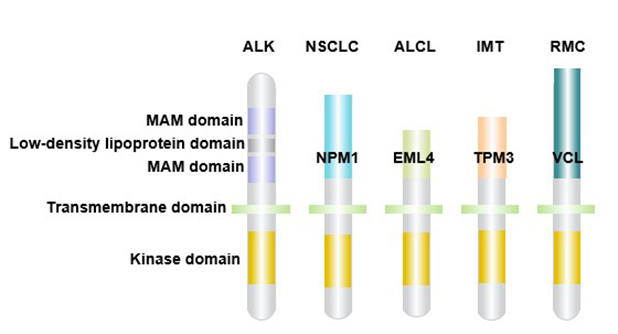
Figure 3 ALK gene rearrangement and cancer
ALCL is a type of malignant T-cell lymphoma expressing CD30. In most cases of ALCL, ALK is activated by rearrangement of chromosomes. NPM1-ALK is the most common translocation in ALCL, which is found in 75% ~ 80% of ALK-positive ALCL patients, followed by TPM3-ALK, which is found in 12% ~ 18% of ALCL patients. Other fusion proteins (TFG-ALK, CLTC1-ALK and ATIC-ALK) appeared less frequently.
Lung cancer is one of the most common malignant tumors in the world, with the mortality ranking the first among all malignant tumors, 85% of which are non-small cell lung cancer (NSCLC).
The ALK gene fusion mutation is a common driver gene for non-small cell lung cancer (NSCLC).Studies have shown that EML4-ALK fusion gene variation exists in non-small cell lung cancer (NSCLC) [8]. EML4-ALK positive patients are mostly young people, and most of them are male patients. Patients who do not smoke or have a history of light smoking have a significantly higher positive rate than smokers [9].
At present, more than ten different EML4-ALK variants have been found in NSCLC, and other ALK-fused proteins, including SEC31A-ALK, HIP1-ALK, KIF5B-ALK and KLC1-ALK, are not common in lung cancer.
Inflammatory myofibroblastic tumors (IMTs) are soft tissue stromal tumors. The TPM3/TPM4-ALK fusion gene is present in IMTs. Other genes fused to ALK (including TPM4, CLTC, CARS, and RANBP2) have a lower incidence in IMTs.
Neuroblastoma is the most common extracranial solid tumor in infants and young children. It has a high degree of malignancy and is difficult to treat.
ALK is a susceptibility gene for clustering and sporadic NB, and participates in the pathogenesis of NB in both point mutation and amplification. Mutations in the ALK gene may be one of the factors in the poor prognosis of NB.
Recent studies have found that ALK abnormalities are also present in some common solid tumors.The study found that EML4-ALK gene fusion exists in both breast cancer and colon cancer [10].
Studies have reported multiple ALK fusions in patients with thyroid cancer (including EML4-ALK, GFPT1-ALK, TFG-ALK, and STRNALK) [11]. At the same time, chimeric ALK protein expression was also found in thyroid cancer patients, and downstream AKT and ERK pathways were activated.
ALK gene amplification has also been found in a variety of tumors. Neuroblastoma cell lines and primary neuroblastoma tissues showed ALK gene amplification, causing ALK protein overexpression and promoting tumor development [12]. Therefore, targeting ALK may be a therapeutic method for ALK-amplified tumors.
ALK gene amplification and ALK gene copy number increase were also found in NSCLC patients. The increase of ALK gene copy number only existed in a small number of cells and was not related to the increase of ALK expression in tissues, and was not an important driving event of tumor [13].
At present, there are three methods for detecting ALK fusion genes in clinical practice: fluorescent in situ hybridization (FISH), immunohistochemistry (IHC) and polymerase chain reaction (PCR) [14].
FISH is the "gold standard" for the detection of ALK fusion gene, with advantages of sensitivity, specificity and good repeatability. FISH method can detect different fusion proteins and variants, and has a good predictive value of efficacy.
With the improvement of sensitivity, IHC technology becomes a feasible screening method [15]. IHC is low cost and easy to operate, but it is difficult to achieve standardization process. Therefore, IHC is suitable as a primary screening method, and FISH can be further used to determine the positive.
The advantage of RT-PCR method is that it has high specificity and can identify various ALK fusion types.
The disadvantage of this method is that it requires high quality of DNA samples and requires fresh or frozen tumor tissues.
Currently, there are some new platforms for ALK fusion gene detection using paraffin-embedded samples.
The abnormality of ALK gene is closely related to the occurrence, development and prognosis of various solid tumors. Targeted treatment of ALK fusion protein has achieved good results in the treatment of various malignant tumors. In addition, since ALK is not widely expressed in adult tissues, the therapeutic toxicity of targeting ALK is small.
ALK inhibitors mainly include crizotinib, ceritinib and erlotinib.
Crazotinib is the first generation of ALK tyrosine kinase inhibition and is an oral small molecule. Crizotinib can act on three targets of ALK, c-MET and ROS1. It is the first ALK inhibitor introduced in the treatment of ALK-dependent NSCLC. Similarly, Crizotinib has shown good efficacy in both ALK-positive IMTs and anaplastic ALCL pediatric patients.
Ceritinib can effectively inhibit ALK mutation after treatment with Crazotinib [16]. Ceritinib is approved as a second-line treatment for ALK-positive NSCLC patients.
Alectinib, a second-generation ALK inhibitor, is 10 times more efficient than crazotinib and can resist most mutations in the ALK kinase region, including L1196M, F1174L, R1275Q and C1156Y [17]. Alectinib has been approved by the FDA and the European Medicines Agency (EMA) as a second-line treatment for ALK-positive NSCLC patients.
For the resistance of ALK inhibitors, new ALK inhibitors, such as Entrectinib, Lorlatinib and Brigatinib, are also under development. Entrectinib, as a multi-target drug, showed strong anti-tumor activity. It is A selective inhibitor of ALK, TPMA, B and C and ROS1, and its efficacy is 36 times higher than that of crizotinib [18].
In addition to directly blocking ALK, there are some drugs that can indirectly target ALK. Heat shock protein is a chaperone protein that stabilizes a variety of proteins including ALK. Inhibition of heat shock protein HSP90 has a certain effect in Crizotinib-resistant tumor models [19].
The common problem with ALK inhibitors is drug resistance. The main drug resistance mechanisms include: primary drug resistance, ALK-related secondary drug resistance, bypass activation, and some rare drug resistance mechanisms.
Primary drug resistance means that the tumor does not respond to initial treatment. The first-line and second-line treatment of ALK-positive advanced NSCLC showed that some patients had primary resistance to Crizotinib. This is also the case with clinical studies of Ceritinib, Erlotinib, and Brigatinib.
It includes: secondary mutations in the ALK kinase domain, ALK gene fusion, and gene amplification [20].
The second mutation in the ALK kinase region is a cluster mutation in the ATP binding site of the kinase domain.
ALK fusion gene amplification refers to a 2-fold increase in intracellular ALK gene after treatment, which ultimately affects drug binding and produces resistance. Ceritinib, Alectinib and Brigatinib all have secondary mutations in the ALK kinase region that lead to drug resistance.
The bypass activation pathway is an ALK independent resistance mechanism. When ALK-dependent signaling pathways are blocked, tumor cells may selectively activate other signaling pathways. Activation of pathways such as EGFR or insulin-like growth factor is involved in the resistance of Crizotinib.
Other rare resistance mechanisms such as Epithelial-mesenchymal transition [21] and ALK gene deletion can cause Crizotinib resistance. Experiments have shown that high expression of P-glycoprotein can cause Crizotinib and Ceritinib resistance [22].
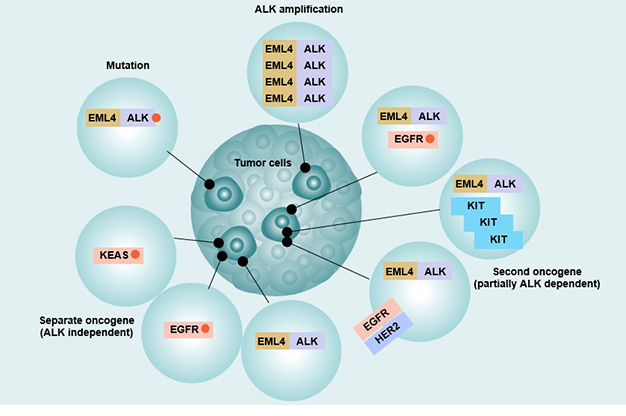
Figure 4 Resistance Mechanism of EML4-ALK
References
[1] Schlessinger J. Cell signaling by receptor tyrosine kinases [J]. Cell, 2000, 141(7): 1117.
[2] Morris S W, Kirstein M N, Valentine M B, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma [J]. Science, 1994,263(5151): 1281-1284.
[3] Li R, Morris S W. Development of anaplastic lymphoma kinase (ALK) small-molecule inhibitors for cancer therapy [J]. Medicinal Research Reviews, 2010, 28(3): 372-412.
[4] Stoica G E, Kuo A, Powers C, et al. Midkine Binds to Anaplastic Lymphoma Kinase (ALK) and Acts as a Growth Factor for Different Cell Types [J]. Journal of Biological Chemistry, 2002, 277(39): 35990-35998.
[5] Chen Z, Sasaki T, Tan X, et al. Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene [J]. Cancer research, 2010, 70(23): 9827-9836.
[6] Bunting S F, Nussenzweig A. End-joining, translocations and cancer [J]. Nature Reviews Cancer, 2013, 13(7): 443-54.
[7] Bridge J A, Kanamori M, Ma Z, et al. Fusion of the ALK Gene to the Clathrin Heavy Chain Gene, CLTC, in Inflammatory Myofibroblastic Tumor [J]. American Journal of Pathology, 2001, 159(2): 411-415.
[8] Soda M, Choi Y L, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer [J]. NATURE, 2007, 448(7153): 561-566.
[9] Kwak E L, Bang Y J, Camidge D R, et al. Anaplastic Lymphoma Kinase Inhibition in Non-Small-Cell Lung Cancer [J]. New England Journal of Medicine, 2010, 363(18): 1693-1703.
[10] Lin E, Li L, Guan Y, et al. Exon Array Profiling Detects EML4-ALK Fusion in Breast, Colorectal, and Non-Small Cell Lung Cancers [J]. Molecular Cancer Research, 2009, 7(9): 1466-1476.
[11] Kelly L M, Barila G, Liu P, et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer [J]. Proceedings of the National Academy of Sciences, 2014, 111(11): 4233-4238.
[12] Osajima-Hakomori Y, Miyake I, Ohira M, et al. Biological Role of Anaplastic Lymphoma Kinase in Neuroblastoma [J]. American Journal of Pathology, 2005, 167(1): 0-222.
[13] Salido M, Pijuan L, Martínez-Avilés L, et al. Increased ALK Gene Copy Number and Amplification are Frequent in Non-small Cell Lung Cancer [J]. Journal of Thoracic Oncology, 2011, 6(1): 21-27.
[14] Rusthoven C G, Doebele R C. Management of Brain Metastases in ALK-Positive Non-Small-Cell Lung Cancer [J]. Journal of Clinical Oncology, 2016: JCO.2016.67.2410.
[15] Minca E C, Portier B P, Wang Z, et al. ALK Status Testing in Non-Small Cell Lung Carcinoma [J]. The Journal of Molecular Diagnostics, 2013, 15(3): 341-346.
[16] Friboulet L, Li N, Katayama R, et al. The ALK Inhibitor Ceritinib Overcomes Crizotinib Resistance in Non-Small Cell Lung Cancer [J]. Cancer Discovery, 2014, 4(6): 662-673.
[17] Kodama T, Tsukaguchi T, Yoshida M, et al. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance [J]. Cancer Letters, 2014, 351(2): 215-221.
[18] Drilon A, Siena S, Ou S H I, et al. Safety and Antitumor Activity of the Multi-Targeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib (RXDX-101): Combined Results from Two Phase 1 Trials (ALKA-372-001 and STARTRK-1) [J]. Cancer Discovery, 2017, 7(4): CD-16-1237.
[19]Sang J, Acquaviva J, Friedland J C, et al. Targeted Inhibition of the Molecular Chaperone Hsp90 Overcomes ALK Inhibitor Resistance in Non-Small Cell Lung Cancer [J]. Cancer Discovery, 2013, 3(4): 430-443.
[20] Doebele R C, Pilling A B, Aisner D L, et al. Mechanisms of Resistance to Crizotinib in Patients with ALK Gene Rearranged Non–Small Cell Lung Cancer [J]. Clinical Cancer Research, 2014, 15(2): 133-135.
[21] Kim H R, Kim W S, Choi Y J, et al. Epithelial-mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4-ALK translocation [J]. Molecular Oncology, 2013, 7(6): 1093-1102.
[22] Katayama R, Sakashita T, Yanagitani N, et al. P-glycoprotein Mediates Ceritinib Resistance in Anaplastic Lymphoma Kinase-rearranged Non-small Cell Lung Cancer [J]. EBioMedicine, 2015, 3(C): 54-66.
[23] Remon J, Besse B. Immune checkpoint inhibitors in first-line therapy of advanced non-small cell lung cancer [J]. Current Opinion in Oncology, 2017, 29(2): 1.


Comments
Leave a Comment