Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
The advent of immune checkpoint inhibitors (ICIs) marks a major breakthrough in cancer treatment, significantly improving outcomes for patients with various cancers by activating the host immune system against tumors [1]. However, ICIs face challenges such as low response rates, immune-related adverse events (IrAEs), and tumor resistance [2-4], prompting continued exploration of new immune checkpoint molecules to optimize treatment strategies [5].
B and T lymphocyte attenuator (BTLA, also known as CD272) is a co-inhibitory receptor discovered in recent years, belonging to the immunoglobulin superfamily. It plays a key role in maintaining immune homeostasis, primarily mediating immune attenuation in B cells and T cells [6,7]. Unlike some co-inhibitory receptors that are upregulated after activation, BTLA is constitutively expressed on resting T cells and shows high expression levels in peripheral lymphoid tissues, suggesting its potential involvement in establishing self-tolerance during early T cell development [8]. BTLA is widely expressed on B cells, CD4+ and CD8+ T cells, and plasmacytoid dendritic cells (pDCs), showing high protein expression in healthy individuals [9].
Aberrant BTLA expression or dysfunction is associated with various diseases. For instance, in chronic lymphocytic leukemia (CLL) patients, BTLA expression is significantly elevated on CD4+ and CD8+ T cell surfaces, and high BTLA levels correlate with a shorter time to initial treatment, suggesting it promotes T cell exhaustion and limits anti-tumor immune responses [10]. Furthermore, functional single nucleotide polymorphisms (SNPs) in the BTLA gene, such as the 590C site, are significantly associated with susceptibility to rheumatoid arthritis (RA). Patients carrying this allele have an earlier disease onset, and this mutant BTLA loses its ability to inhibit IL-2 production in Jurkat T cells, further confirming its role in autoimmune protection [11]. In systemic lupus erythematosus (SLE), HVEM protein levels are decreased while BTLA remains relatively stable, suggesting that BTLA agonists might offer a new therapeutic approach for SLE patients with low HVEM expression [9].
Given the central role of BTLA in immune regulation and disease progression, in-depth research into its mechanisms and clinical applications is of great value. This article aims to comprehensively review the molecular background, mechanisms of action, signaling pathways, associated diseases, and drug development progress related to BTLA, and to prospect its potential in the field of immunotherapy.
BTLA belongs to the B7-CD28 family of inhibitory immune checkpoint receptors. Its inhibitory function primarily relies on the immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM) contained within its intracellular segment. After tyrosine phosphorylation, these motifs can specifically recruit the SH2 domain-containing phosphatases SHP1 and SHP2, thereby initiating negative signal transduction [13,14].
The ITIM motif of BTLA plays a dominant role in recruiting SHP1, particularly through high-affinity binding with its N-terminal SH2 domain (nSH2) [13]. Unlike PD-1, which primarily recruits SHP2 via its ITSM motif binding the C-terminal SH2 domain (cSH2) of SHP2, the molecular volume of the first amino acid following the phosphorylated tyrosine in BTLA's ITIM exhibits a "bell-shaped" dependency for SHP1 recruitment. Structural differences determine its effector protein selectivity [13]. Mutating the intracellular tyrosine motifs of BTLA reduces SHP-1/2 recruitment while preserving Grb2 binding capacity, thereby enhancing the NFAT signaling pathway and improving T cell persistence and anti-tumor function in vivo [12]. These findings reveal that the structure of BTLA's intracellular motifs is decisive for its inhibitory function and therapeutic development.
BTLA is highly expressed on B cells, CD4+ and CD8+ T cells, and plasmacytoid dendritic cells (pDCs) in healthy individuals [9], indicating its broad role in immune homeostasis. In disease states, BTLA expression patterns change. For example, in non-small cell lung cancer (NSCLC), high BTLA expression on tumor cells is associated with lymph node invasion, advanced stage, and poor relapse-free survival (RFS) [15]. Significant BTLA overexpression has also been observed in lung adenocarcinoma [5].
During T cell development, BTLA expression is low in the thymus but significantly increased in peripheral T cells, showing a negative correlation with CD5 expression [8]. BTLA regulates CD5 levels in CD4 T cells through its own signaling (independent of the HVEM ligand), which is crucial for establishing self-tolerance in newly generated T cells. BTLA-deficient recent thymic emigrants (RTEs) can trigger multi-organ autoimmune disease, highlighting its role in peripheral tolerance [8].
Epigenetic regulation also influences BTLA expression. Hypomethylation of promoter CpG sites (e.g., cg24157392, cg03995631) is associated with upregulated BTLA mRNA and protein expression, predicting longer overall survival (OS) and higher levels of immune cell infiltration in melanoma patients, and can serve as a biomarker for immunotherapy response and prognosis [16]. Additionally, the T cell lineage protein THEMIS acts as a "rheostat" for BTLA signaling, enabling T cells to resist BTLA-mediated inhibition and promoting T cell development and maintenance [17], demonstrating fine-tuning of immune checkpoint function by intracellular factors.
The primary physiological ligand for BTLA is Herpesvirus Entry Mediator (HVEM, also known as TNFRSF14), which belongs to the tumor necrosis factor receptor (TNFR) family [18-20]. The binding of BTLA to HVEM constitutes a critical inhibitory immune checkpoint, regulating T cell and B cell immune responses, and its blockade is considered a new strategy for cancer treatment [21].
HVEM, as a multifunctional receptor, can also bind other ligands such as LIGHT (TNFSF14) and CD160, forming a network of co-stimulatory or co-inhibitory signals [22,20,23]. In rheumatoid arthritis (RA) patients, HVEM and LIGHT expression on T lymphocytes is decreased, while BTLA is increased, suggesting the involvement of this pathway in RA pathogenesis [24]. CD160 gene polymorphisms are also associated with susceptibility to autoimmune thyroid diseases (AITD) like Graves' disease, underscoring the critical role of the CD160/HVEM/LIGHT/BTLA pathway in autoimmunity [25].
The BTLA-HVEM axis can mediate bidirectional signaling: BTLA delivers an inhibitory signal to the T cell, while HVEM transmits a signal into the cell expressing it [18,22]. Both molecules can bind in cis on the same cell surface, potentially blocking trans binding of other ligands to HVEM and reducing co-stimulatory activity, while the inhibitory function of BTLA is retained [22]. Trans binding refers to the interaction between BTLA and HVEM on different cells and is considered the primary inhibitory pathway [21]. Quantitative detection methods for the HVEM-BTLA cis complex have been established, providing tools for further study of its functional regulation [18].
In a hepatobiliary injury model, HVEM- or BTLA-deficient mice exhibited more severe injury and impaired repair after DDC induction, accompanied by dysregulated gut microbiota and IgA responses, suggesting this signaling axis limits liver injury by regulating the gut microbiome [26]. In anti-viral immunity, the HVEM-BTLA bidirectional co-stimulatory system drives memory CD8 T cell differentiation, and its deficiency impairs effector CD8 T cell survival and immune memory formation [23].
In the tumor microenvironment, melanoma-associated fibroblasts (MAFs) upregulate inhibitory receptors including HVEM, suppressing CTL activity [27]. Anti-HVEM monoclonal antibodies show anti-tumor effects both in vitro and in vivo, enhancing T cell activation and reducing exhaustion [19,20,28]. In CAR-T therapy, knocking out BTLA reduces SHP-1/2 recruitment and enhances the anti-tumor function of CAR-T cells [12], highlighting the significant value of the BTLA-HVEM axis as a therapeutic target.
BTLA ligand binding properties show evolutionary diversity. In zebrafish, research has identified a PD-L1/BTLA co-inhibitory axis, potentially serving as an evolutionary alternative to the mammalian PD-L1/PD-1 axis [29]. During Edwardsiella tarda infection, zebrafish PD-L1 (DrPD-L1) and BTLA (DrBTLA) are upregulated on MHC II+ macrophages and CD8+ T cells, respectively, and exhibit high affinity for each other (KD = 5.68 nM). Blocking this interaction enhances the killing of infected macrophages by CD8+BTLA+ T cells and reduces pathogen immune escape [29]. This primitive checkpoint axis regulates CD8+ T cell activation in teleost fish, demonstrating the ancient evolutionary roots of BTLA in immune regulation.
Although PD-1 was recently thought to exist only in tetrapods, database analyses suggest its presence in bony and cartilaginous fish as well [30]. The conserved extracellular and intracellular binding patterns and glycosylation sites of PD-1 and its ligands during evolution support their ancient origin, indicating the conservation and adaptability of immune checkpoint mechanisms across species.
Upon T cell activation, the intracellular ITIM and ITSM motifs of BTLA undergo tyrosine phosphorylation, subsequently recruiting the phosphatases SHP1 and SHP2, which inhibit early T cell activation and function [12,14,31]. Unlike PD-1, which preferentially recruits SHP2 via ITSM, BTLA primarily recruits SHP1 through its ITIM motif [13]. Structural studies show that the molecular volume of the first residue following the phosphorylated tyrosine in the ITIM has a bell-shaped dependency for SHP1 recruitment; BTLA has an alanine at this position, favoring stable binding with the nSH2 domain of SHP1 [13]. PD-1, with a glycine at this position, has a weaker ITIM role in SHP1 recruitment; replacing it with alanine enhances interaction with SHP1 [13]. PD-1 recruitment of SHP2 requires the participation of both ITIM and ITSM, binding the nSH2 and cSH2 domains of SHP2 respectively, inducing conformational changes that activate SHP2 phosphatase activity [32,33]. Although SHP1 and SHP2 show functional redundancy in inhibiting the differentiation of naive T cells into effector and memory phenotypes, SHP1 generally plays a dominant role [34]. The differences in SHP enzyme recruitment between BTLA and PD-1 reflect the precise regulation of downstream signaling pathways by immune checkpoint receptors.
By recruiting SHP1 and SHP2, BTLA dephosphorylates key molecules in the T cell receptor (TCR) signaling pathway, such as CD3ζ, ZAP70, and Lck, blocking early TCR signals and inhibiting T cell activation, proliferation, and cytokine production [17]. In an asthma model, a BTLA agonist inhibited NF-κB signaling in a SHP-1-dependent manner, reduced Th17 cell numbers and IL-17 levels, and alleviated airway hyperresponsiveness and lung inflammation [35]. High BTLA expression on T cells from CLL patients leads to decreased IL-2 and IFN-γ production, limiting anti-tumor responses [10]. BTLA gene polymorphisms (e.g., the 590C SNP) can impair its ability to inhibit IL-2 production in Jurkat T cells, contributing to autoimmune disease development [11].
Regarding T cell differentiation, SHP1 and SHP2 jointly inhibit the differentiation of naive T cells into effector and central memory phenotypes [34]. BTLA, through its signaling early in T cell development, regulates CD5 expression. Increased CD5 levels are associated with enhanced self-peptide-MHC recognition, indicating BTLA's role in establishing self-tolerance in newly generated T cells [8]. BTLA-deficient RTEs cause multi-organ autoimmunity dependent on CD4 T cells and MHC class II molecules [8]. In anti-viral immunity, BTLA expressed on CD8α+ dendritic cells can act as a trans-activating ligand, delivering a co-stimulatory signal via HVEM on T cells to drive memory CD8 T cell differentiation and survival [23], demonstrating the critical role of the BTLA-HVEM axis in immune memory.
BTLA is also expressed on B cells, NK cells, and macrophages, participating in the regulation of their functions. In colorectal cancer (CRC), BTLA expression correlates with the infiltration levels of naive B cells, memory B cells, etc., suggesting its potential role in B cell-mediated immunity [36]. Although direct evidence is scarce, it is speculated that BTLA might inhibit B cell activation, proliferation, and antibody secretion through mechanisms similar to those in T cells. In CLL, BTLA dysregulation primarily affects T cells; its direct effects on B cells themselves require further exploration [10].
In a glomerulonephritis model, BTLA exerted a protective effect on the kidneys by inhibiting pathogenic T cells and promoting regulatory T cell (Treg) expansion [37]. In CRC, BTLA expression is associated with resting NK cell infiltration and is involved in NK cell-mediated cytotoxicity pathways [36]. Infiltration of monocytes, M0, and M1 macrophages is also correlated with BTLA expression, suggesting BTLA might influence the tumor microenvironment by regulating macrophage polarization and function [36]. The specific mechanisms by which BTLA directly regulates these cells remain a key focus for future research.
BTLA, together with PD-1, CTLA-4, TIGIT, LAG-3, TIM-3, and other immune checkpoints, forms a complex regulatory network. In Sézary syndrome, tumor T cells show upregulated BTLA, FCRL3, and TIGIT expression, while LAG-3+ cells are decreased, indicating heterogeneous expression patterns of different receptors in the disease [38]. In NSCLC, BTLA, LAG-3, TIGIT, CTLA-4, PD-1, etc., are generally upregulated, but tumor subpopulations show different immune checkpoint gene expression profiles, suggesting heterogeneity in immune escape mechanisms [5]. In biliary tract cancer (BTC) patients, a high frequency of CD8+BTLA+ T cells is associated with better overall survival, while changes in CD4+TIM3+ and CD8+VISTA+ T cells correlate with different clinical outcomes, highlighting the unique prognostic roles of individual checkpoints [39].
Combined targeting of multiple checkpoints can produce synergistic effects. In a mouse islet transplant model, the combination of CTLA4Ig and an anti-BTLA monoclonal antibody achieved long-term graft survival and induced donor-specific tolerance [40]. Engineered dendritic cells (DCs) co-expressing CTLA-4, PD-1, and BTLA ligands effectively suppressed CD4+ T cell proliferation and pro-inflammatory cytokines, increased Treg frequency, and suppressed autoimmune thyroiditis [41]. In the tumor microenvironment, MAFs upregulate TIGIT and BTLA via arginase activity, inhibiting CTL function [27]; CD47 blockade can reduce exhausted BTLA+ CD4+ T cells and promote NK and CD8+ T cell expansion [42]. At the signaling level, T cell-specific deletion of both SHP1 and SHP2, while inhibiting naive T cell differentiation, led to poor tumor control and unresponsiveness to PD-1 blockade, revealing the critical role of SHP1/SHP2 in maintaining T cell survival and tumor immunity [34]. These studies collectively indicate complex cross-regulation between BTLA and other immune checkpoints, and a deep understanding of their interactions is crucial for developing combination immunotherapies.
BTLA expression is regulated at multiple levels, including epigenetic mechanisms, intracellular factors, and microRNAs. Hypomethylation of specific CpG sites in the promoter region is associated with high BTLA mRNA and protein expression, predicting better prognosis and higher immune cell infiltration in melanoma patients [16]. CD5 and BTLA show negative correlation in expression in the thymus and periphery; BTLA regulates CD5 levels through its own signaling, participating in the establishment of self-tolerance [8]. THEMIS acts as a "rheostat" for BTLA signaling, limiting its inhibitory effect and promoting T cell development and maintenance [17]. In CLL, miR-155-5p upregulation can partially reduce BTLA protein levels in B cells, but silencing miR-155-5p in T cells did not significantly alter BTLA expression, suggesting cell-type-specific regulation and providing a basis for immunotherapy targeting miR-155-5p [43].
BTLA is highly expressed in various tumors and is associated with poor patient prognosis and immune escape. Pan-cancer analysis shows that approximately 19% of tumors have high BTLA RNA expression, which is independently correlated with checkpoints like PD-1, CTLA-4, HVEM, and CD160 [44]. In osteosarcoma, soluble BTLA (sBTLA) is significantly associated with the risk of lung metastasis and disease progression, showing biomarker potential [45].
In non-small cell lung cancer (NSCLC), high BTLA expression on tumor cells is associated with lymphatic invasion, advanced stage, and poorer relapse-free survival (RFS) and overall survival (OS) [15]. BTLA expression is upregulated in oral squamous cell carcinoma (OSCC), positively correlating with PD-1, PD-L1, PD-L2, CD96, etc., forming a local immune checkpoint network [47]. High HVEM and BTLA mRNA expression in prostate cancer predicts poorer progression-free survival [19]. Increased BTLA expression on T cells in CLL patients, and high BTLA on CD4+ T cells, correlates with a shorter time to initial treatment [10]. In advanced-stage mycosis fungoides (MF), the density of exhausted BTLA+ CD4+ T cells increases around tumors, forming an inhibitory microenvironment [42]. In Sézary syndrome (SS), tumor T cells show upregulated BTLA, FCRL3, and TIGIT expression [38]. In biliary tract cancer (BTC), a high frequency of CD8+BTLA+ T cells is associated with better overall survival, demonstrating the complexity of BTLA's role across different tumor contexts [39].
In colorectal cancer (CRC), BTLA expression in tumor tissue is lower than in normal tissue, and low BTLA is associated with poorer overall survival, classifying it as a favorable prognostic factor [36]. In melanoma, BTLA promoter hypomethylation leads to high expression, predicting longer overall survival and better response to immunotherapy [16], contrasting with the conventional view of BTLA as an inhibitory molecule and suggesting its function is tumor-type specific.
BTLA plays a key role in maintaining autoimmune tolerance. Its gene polymorphisms, abnormal expression, or functional defects are associated with various autoimmune diseases.
In rheumatoid arthritis (RA), the BTLA gene 590C SNP is associated with disease susceptibility and earlier onset. This mutant BTLA loses its ability to inhibit IL-2 production in vitro [11]. AI analysis indicates that BTLA interacts with a network of RA candidate genes including PADI4, FCGR3, TNFRSF1B, and ITGAV [50]. Although the proportion of circulating T cells expressing BTLA is increased in RA patients, HVEM and LIGHT expression are decreased [24], suggesting that an imbalance in the BTLA-HVEM-LIGHT pathway might lead to insufficient effective immune suppression, contributing to disease progression.
In systemic lupus erythematosus (SLE) patients, BTLA protein levels are comparable to healthy individuals, but HVEM protein is significantly reduced, and HVEM gene expression negatively correlates with disease activity [9], providing a rationale for BTLA agonist therapy in SLE patients with low HVEM expression.
In Sjögren's syndrome (SjS) patients, the expression levels and co-expression frequencies of BTLA, HVEM, and CD160 on peripheral blood lymphocytes are decreased. Co-expression of BTLA/HVEM and CD160/HVEM on T cells is reduced, indicating dysregulation of this axis in SjS, potentially making it a therapeutic target [51]. Earlier studies found no association between the BTLA 590C SNP and susceptibility to SLE or SjS [11], suggesting different modes of action for BTLA gene polymorphisms in different autoimmune diseases.
Ankylosing spondylitis (AS) is associated with the BTLA gene rs2171513 polymorphism in the Chinese Han population [54]. Autoimmune thyroid diseases (AITD) like Graves' disease are associated with the CD160 gene rs744877 polymorphism, highlighting the role of the CD160/HVEM/LIGHT/BTLA pathway in autoimmunity [25].
In atherosclerosis, treatment with a BTLA agonist antibody (3C10) alleviated lesions, reduced follicular B2 cells, increased regulatory B and T cells, and enhanced plaque collagen content, suggesting a plaque-stabilizing effect [55]. In experimental glomerulonephritis, BTLA deficiency worsened nephritis, while a BTLA agonist antibody alleviated kidney inflammation and protected renal function by inhibiting pathogenic Th1 cells and promoting Treg expansion [56,37]. These findings indicate the significance of BTLA signaling in limiting kidney inflammation, and antibody-mediated modulation of BTLA might become a therapeutic strategy for human glomerulonephritis.
In HTLV-1 infection, the viral protein HBZ suppresses BTLA and LAIR-1 expression on infected cells and ATL cells, while enhancing TIGIT and PD-1 expression but impairing their inhibitory function [52]. HBZ interacts with THEMIS, hindering SHP-2 co-localization with PD-1 and weakening PD-1 and TIGIT signaling, thereby promoting T cell proliferation [52]. Sera from HAM/TSP patients and HTLV-1-derived extracellular vesicles (EVs) and exosomes show elevated levels of soluble immune checkpoint molecules like sBTLA, LAG-3, and PD-L2 [49]. These exosomes carrying BTLA can impair healthy CD8 T cell function [49]. HBZ knockdown reduces EV release and immune checkpoint molecule secretion, suggesting its role in HTLV-1 neuroinflammation via exosome-mediated immune regulation [49].
In an asthma model, BXD75 mice exhibited neutrophil skewing, steroid resistance, and elevated Th17 cells. CD4+ T cells in the lungs showed increased HVEM expression and decreased BTLA, along with enhanced NF-κB signaling [35]. A BTLA agonist alleviated airway hyperresponsiveness and lung inflammation, and in vitro, it inhibited NF-κB via SHP-1 mediation, reducing Th17 cells and IL-17 [35], suggesting BTLA agonists could be used for steroid-resistant asthma.
The PD-L1/BTLA checkpoint axis identified in zebrafish regulates CD8+ T cell activation during Edwardsiella tarda infection. Blocking this axis enhances the killing of infected macrophages by CD8+BTLA+ T cells and reduces pathogen escape [29], indicating BTLA's involvement in anti-infection immunity early in evolution.
The immunosuppressed state in critically ill patients is associated with increased HVEM/BTLA co-expression. HVEM+BTLA+ co-expression on CD3+ lymphocytes is elevated in mouse models and patient circulating lymphocytes, positively correlating with TNF-α levels, suggesting its involvement in the development of immunosuppression [48]. HVEM expression can enhance the immunosuppressive and osteogenic capabilities of allogeneic bone marrow-derived mesenchymal stem cells (allo-MSCs), promoting new bone formation in a femoral defect model [31]. HVEM-BTLA signaling limits liver injury in DDC-induced models by regulating gut microbiota and IgA responses [26]. These studies reveal the broad regulatory functions of BTLA in infection, inflammation, and various other physiological and pathological processes.
Soluble BTLA (sBTLA), as a circulating biomarker, shows potential in the diagnosis and prognosis assessment of tumors and other diseases. Plasma sBTLA levels are lower in basal cell carcinoma (BCC) patients compared to healthy controls, while sCTLA-4, sLAG-3, sPD-1, sPD-L1, sTIM-3, etc., are elevated [4], suggesting a unique role for sBTLA in the BCC immune microenvironment.
In osteosarcoma (OS), sBTLA, along with sPDL2 and sCD27, is associated with the risk of lung metastasis, while sBTLA and sTIM3 are associated with the risk of disease progression [45]. Immune subtypes based on sBTLA, sPD-1, sTIM-3, and sPDL2 can distinguish patients with different progression-free survival (PFS) and lung metastasis-free survival (LMFS), suggesting the value of sBTLA in OS prognosis and guiding immunotherapy.
In NSCLC patients receiving chemotherapy or PD-1 blockade therapy, plasma levels of sBTLA and other soluble immune checkpoint molecules (e.g., sCD27, sCD28, sPD-1, sPD-L1) are elevated [46], reflecting tumor burden or therapy-induced immune responses. sBTLA, as part of a multi-marker panel, has potential in assessing NSCLC invasion risk [3]. Cell surface BTLA in NSCLC is associated with lymphatic infiltration, advanced stage, high PD-L1 expression, and poor prognosis [15], suggesting that soluble and membrane-bound forms of BTLA might provide complementary prognostic information.
In biliary tract cancer (BTC), a high frequency of CD8+BTLA+ T cells is associated with better overall survival [39], indirectly supporting the research value of sBTLA in this disease. High HVEM and BTLA mRNA expression in prostate adenocarcinoma is associated with poorer progression-free survival [19], corroborating the role of this axis in tumor progression.
The heterogeneous changes in sBTLA levels and their prognostic associations across different tumors indicate its potential as a single or combined biomarker, providing liquid biopsy information for early cancer diagnosis, progression monitoring, treatment response assessment, and personalized immunotherapy. The specific mechanisms, optimal detection timing, and complexity of interactions with membrane-bound BTLA still require in-depth study.
As an emerging target of great interest in the field of tumor immunotherapy in recent years, BTLA-targeting drugs, particularly inhibitors blocking its interaction with its ligand HVEM, have shown potential in clinical studies for various tumors and also demonstrate application prospects in autoimmune diseases and transplant rejection. Current R&D of BTLA-targeting drugs is highly concentrated in the field of malignant tumors and has entered the mid-to-late stages of clinical research. Some pipelines under investigation are listed in the table below:
| Drug | Mechanism of Action | Drug Type | Investigational indication (disease name) | Investigational institutions | Highest research and development stage |
|---|---|---|---|---|---|
| Recombinant humanized anti-BTLA monoclonal antibody (Shanghai Junshi Biosciences) | BTLA inhibitor | Monoclonal antibody | Classical Hodgkin lymphoma | Refractory classical Hodgkin lymphoma | Extensive-stage small cell lung cancer ect. | Shanghai Junshi Biosciences Co., Ltd. | Shanghai Pulmonary Hospital | Clinical 3 |
| GS-0272 | BTLA inhibitor | Small molecule drug | Rheumatoid arthritis | Gilead Sciences, Inc. | Clinical Phase 1 |
| Polyethylene Glycol-Recombinant Human Endostatin for Injection (Xianhong Pharmaceutical) | BTLA agonist | Recombinant protein | Metastatic non-small cell lung cancer | Shandong Simcere Biopharmaceutical Co., Ltd. | China Pharmaceutical University | Jiangsu Simcere Pharmaceutical Co., Ltd | Clinical Phase 1 |
| ANB-032 | BTLA agonist | Monoclonal antibody | Atopic dermatitis | AnaptysBio, Inc. | Clinical Phase 1 |
| HFB-200603 | BTLA inhibitor | Monoclonal antibody | Late-stage malignant solid tumors | Colorectal cancer | Melanoma | Non-small cell lung cancer, etc. | Gao Cheng Biopharma (Hangzhou) Co., Ltd. | Hifibio SAS | Clinical Phase 1 |
| MB-272 | BTLA inhibitor | Monoclonal antibody | Autoimmune disease | MiroBio Ltd. | Clinical Phase 1 |
| HFB-200604 | BTLA agonist | Monoclonal antibody | Autoimmune disease | Hocheng Biopharma (Hangzhou) Co., Ltd | Clinical application approval |
| MG-B-28 | BTLA inhibitor | Small molecule drug | Tumor | Weill Cornell Medicine | Mansoura University | Preclinical |
(Data as of November 13, 2025, sourced from synapse)
BTLA, as a key co-inhibitory receptor, plays a central role in maintaining T cell self-tolerance, regulating immune responses, and inhibiting T cell function, possessing dual regulatory potential in autoimmune diseases and tumor immunity. sBTLA is an emerging biomarker showing initial value in tumor prognosis and treatment guidance. The development of agonists and antagonists targeting the BTLA/HVEM axis provides new directions for the treatment of autoimmune diseases and tumors.
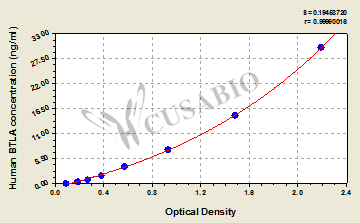
CUSABIO provides BTLA recombinant proteins, antibodies, and ELISA kits to assist your related mechanism research and targeted drug development.
● BTLA Recombinant Protein
Recombinant Human B- and T-lymphocyte attenuator (BTLA), partial (Active); CSB-MP773799HU
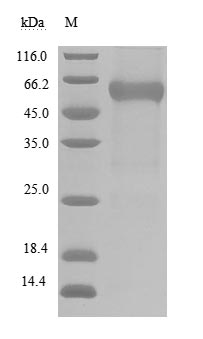
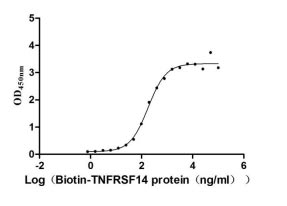
Recombinant Human B- and T-lymphocyte attenuator(BTLA), partial (Active); CSB-MP773799HU1
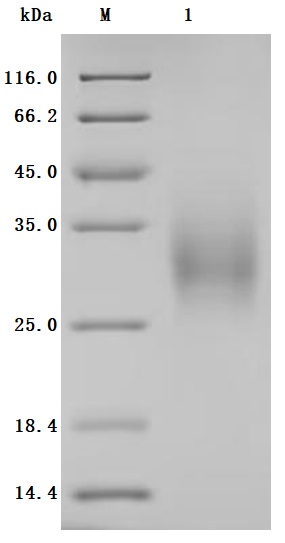
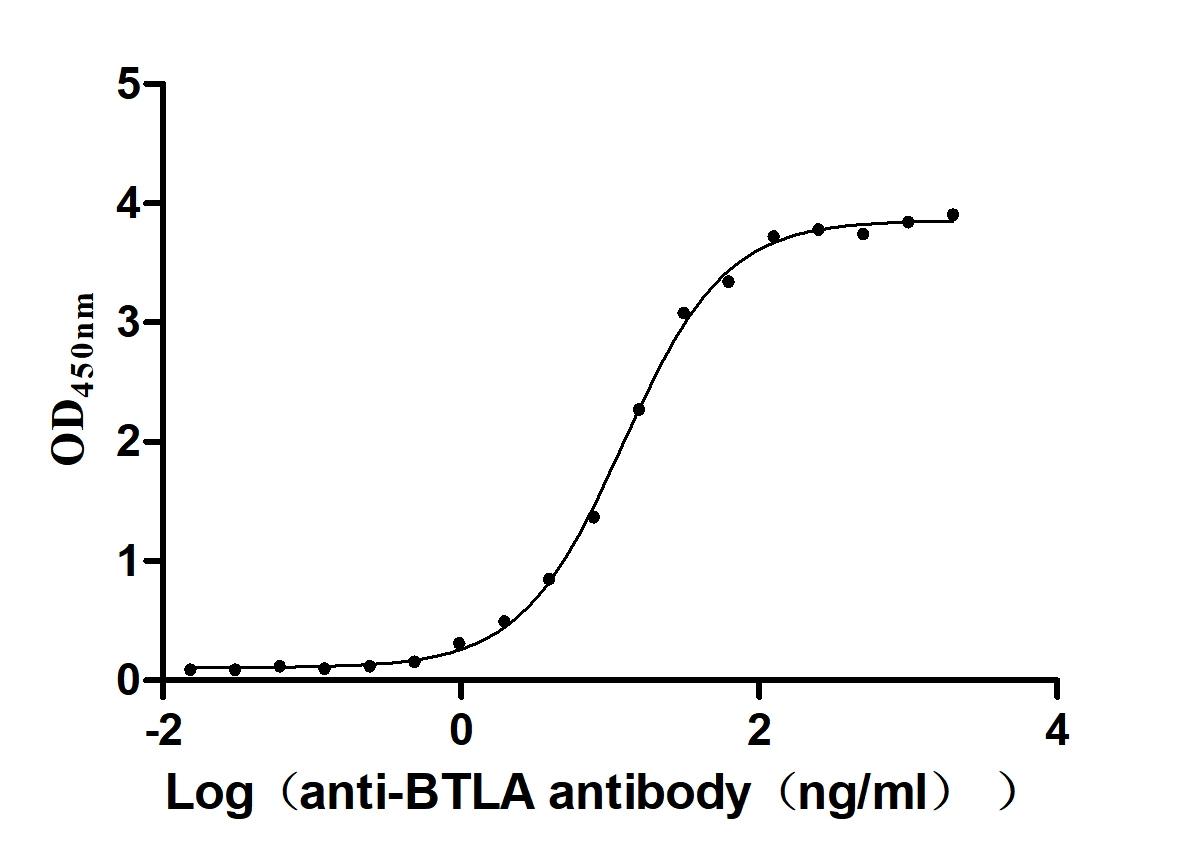
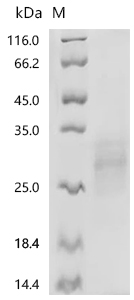
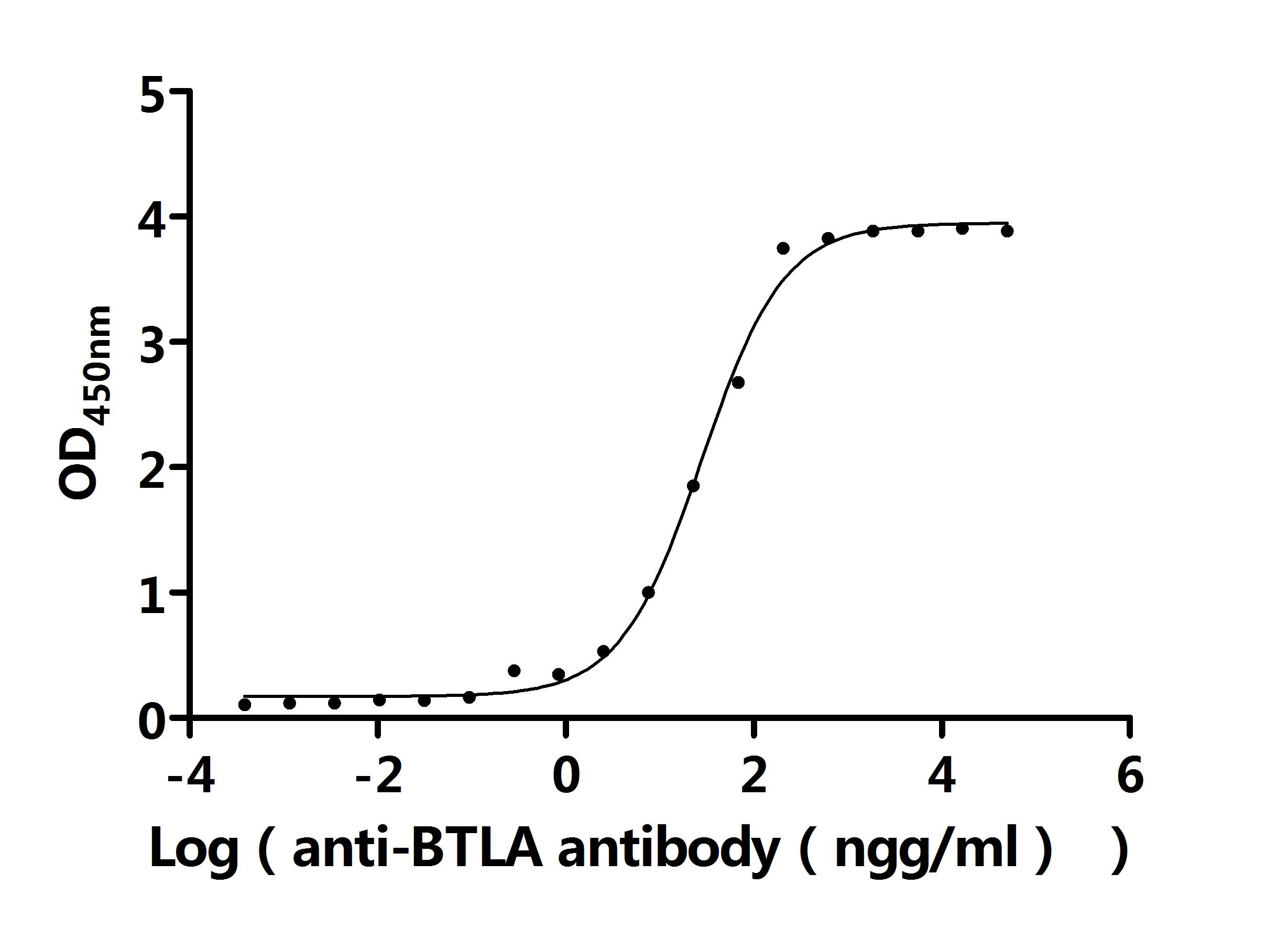
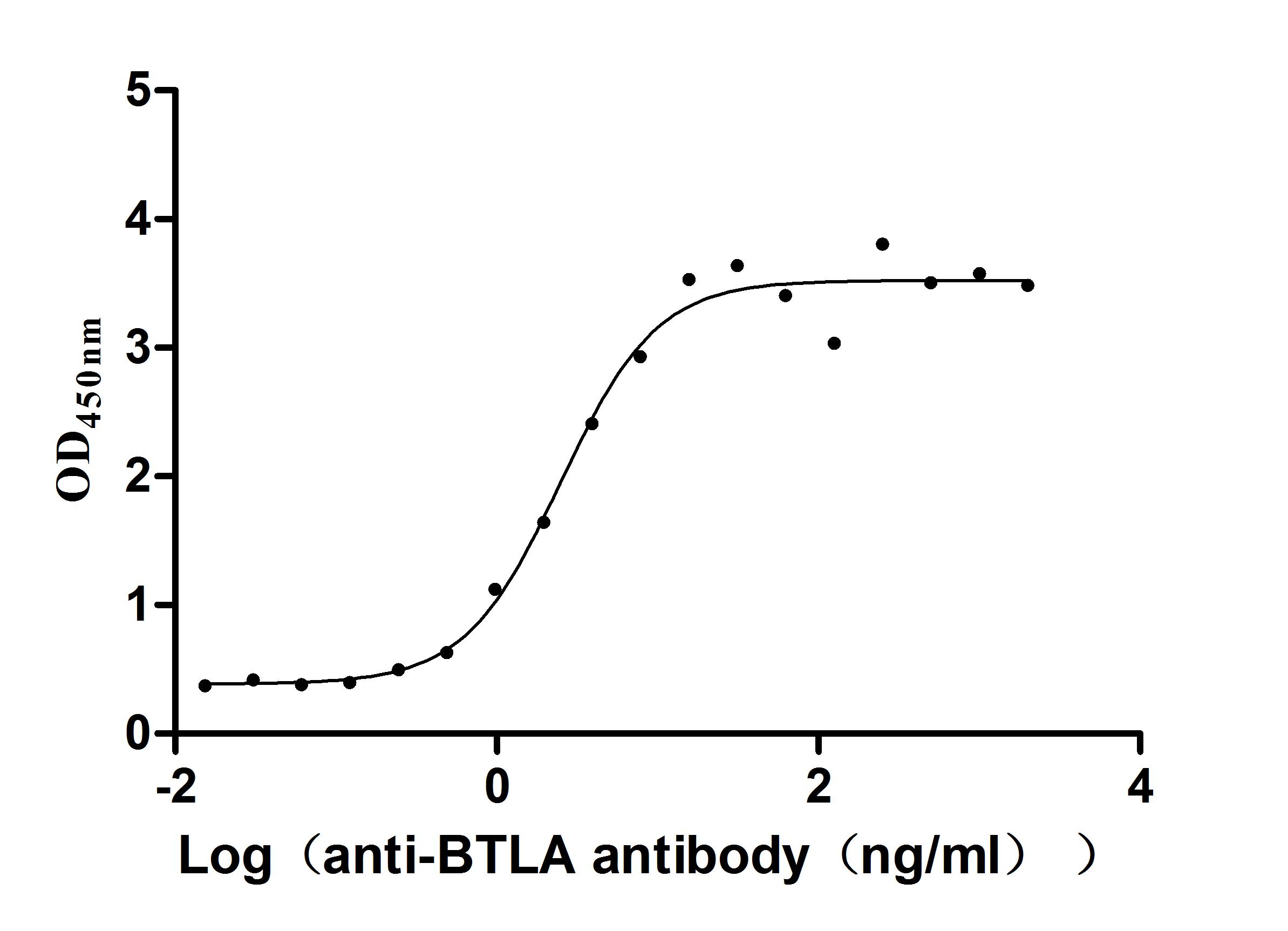
● BTLA Antibodies
BTLA Recombinant Monoclonal Antibody; CSB-RA773799MA1HU
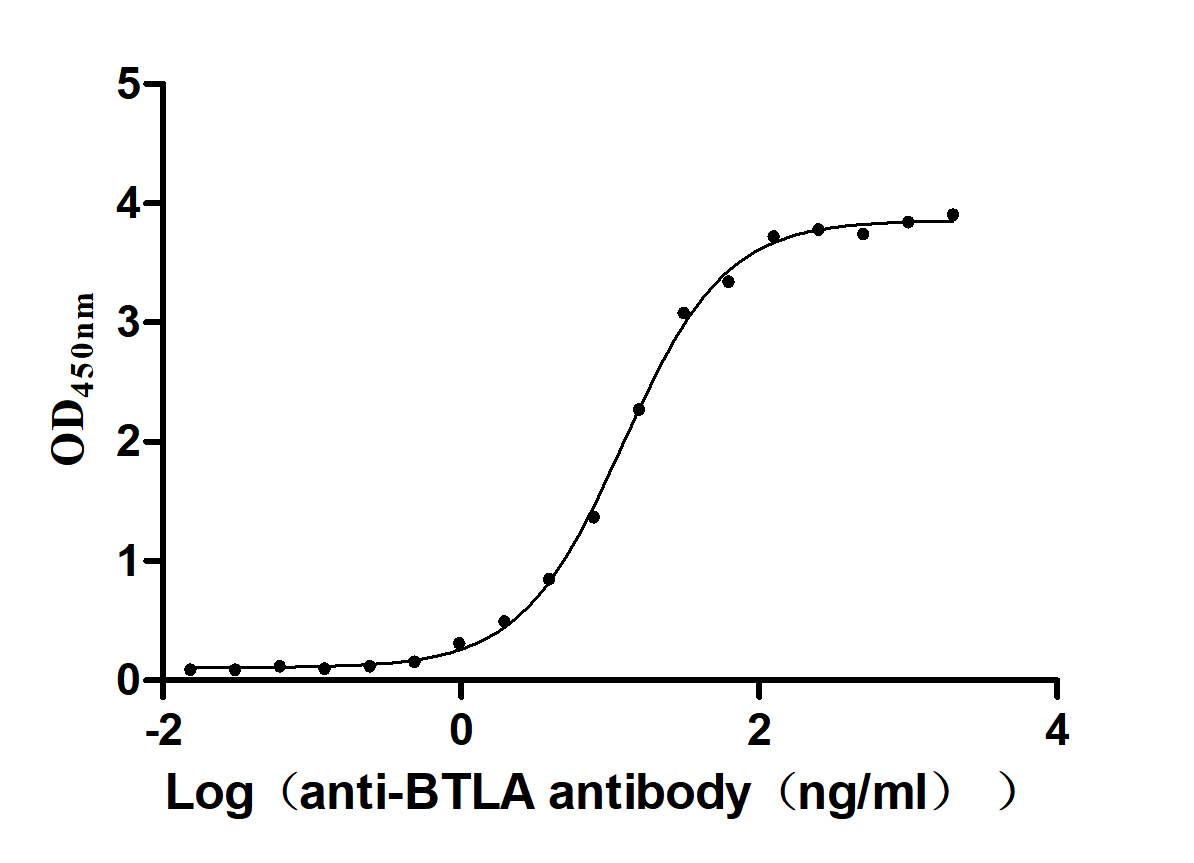
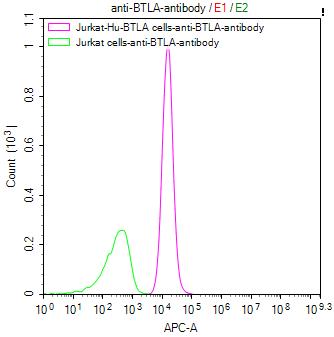
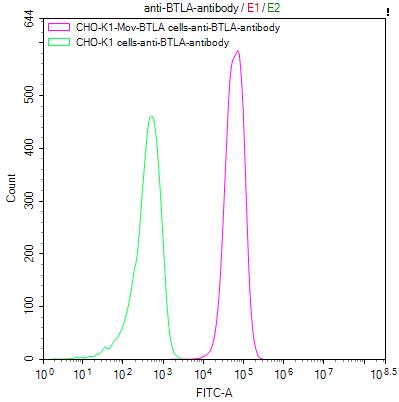
● BTLA ELISA Kit
References
[1] Michael Shapiro, Herut Dor, Anna Gurevich-Shapiro, Tal Etan, Ido Wolf.(2024). Institutional-Level Monitoring of Immune Checkpoint Inhibitor IrAEs Using a Novel Natural Language Processing Algorithmic Pipeline.
[2] Kamran Kaveh, Feng Fu.(2021). Immune checkpoint therapy modeling of PD-1/PD-L1 blockades reveals subtle difference in their response dynamics and potential synergy in combination.
[3] Qinchuan Wang, Yue He, Wanlu Li, Xiaohang Xu, Qingfeng Hu, Zilong Bian, A. Xu, H. Tu, Ming Wu, Xifeng Wu.(2022). Soluble Immune Checkpoint-Related Proteins in Blood Are Associated With Invasion and Progression in Non-Small Cell Lung Cancer.
[4] B. Rapoport, R. Anderson, N. Malinga, H. Steel, P. Meyer, T. Smit, M. Kgokolo.(2023). Transforming growth factor-b1 and soluble co-inhibitory immune checkpoints as putative drivers of immune suppression in advanced basal cell carcinoma.
[5] A. Desai, V. Subbiah, A. Dimou, J. Deshane, Kayla F. Goliwas, S. Ponnazhagan, D. Das, M. Khalil, Y. Lo, Edwin Lin.(2023). Exploring the potential of combination immune checkpoint strategies in non-small cell lung cancer (NSCLC).
[6] Chun Zeng, Tinghe Wu, Y. Zhen, X. Xia, Yong Zhao.(2005). BTLA, a new inhibitory B7 family receptor with a TNFR family ligand.
[7] W. Hobo, W. J. Norde, N. Schaap, H. Fredrix, F. Maas, Karen Schellens, J. Falkenburg, A. Korman, D. Olive, R. van der Voort, H. Dolstra.(2012). B and T Lymphocyte Attenuator Mediates Inhibition of Tumor-Reactive CD8+ T Cells in Patients After Allogeneic Stem Cell Transplantation.
[8] Adeolu O. Adegoke, G. Thangavelu, Ting-Fang Chou, Marcos I. Petersen, K. Kakugawa, J. May, K. Joannou, Qingyang Wang, K. K. Ellestad, Louis Boon, Peter A. Bretscher, H. Cheroutre, Mitchell Kronenberg, T. Baldwin, Colin C. Anderson.(2024). Internal regulation between constitutively expressed T cell co-inhibitory receptors BTLA and CD5 and tolerance in recent thymic emigrants.
[9] Andrew C Vendel, L. Jaroszewski, Matthew D Linnik, Adam Godzik.(2024). B‐ and T‐Lymphocyte Attenuator in Systemic Lupus Erythematosus Disease Pathogenesis.
[10] Christian Sordo-Bahamonde, Seila Lorenzo-Herrero, Alejandra G Martinez-Perez, A. P. Gonzalez-Rodriguez, Á. Payer, E. González-García, Candelaria Aguilar-García, Sara González-Rodríguez, A. López-Soto, A. García-Torre, S. González.(2023). BTLA dysregulation correlates with poor outcome and diminished T cell-mediated antitumor responses in chronic lymphocytic leukemia.
[11] Mie Oki, N. Watanabe, T. Owada, Yoshihiro Oya, K. Ikeda, Y. Saito, R. Matsumura, Y. Seto, I. Iwamoto, H. Nakajima.(2011). A Functional Polymorphism in B and T Lymphocyte Attenuator Is Associated with Susceptibility to Rheumatoid Arthritis.
[12] P. Guruprasad, A. Carturan, Yunlin Zhang, K. G. Kumashie, Ivan J Cohen, G. Ghilardi, Ki-Hyun Kim, Jong-Seo Lee, Yoon Lee, Jong-Hoon Kim, J. Chung, Maksim Shestov, R. Pajarillo, Jaryse Harris, Yong Gu Lee, Michael Wang, H. Ballard, Aasha Gupta, O. Ugwuanyi, S. Hong, Linhui Chen, L. Paruzzo, Shane C Kammerman, R. Patel, O. Shestova, L. Vella, S. Schuster, J. Svoboda, P. Porazzi, M. Ruella.(2023). Modulation of the Btla-HVEM Axis to Enhance CAR T Cell Immunotherapy Against Cancer.
[13] Xiaozheng Xu, T. Masubuchi, Qixu Cai, Yunlong Zhao, E. Hui.(2021). Molecular features underlying differential SHP1/SHP2 binding of immune checkpoint receptors.
[14] J. Chemnitz, R. Parry, K. Nichols, C. June, J. Riley.(2004). SHP-1 and SHP-2 Associate with Immunoreceptor Tyrosine-Based Switch Motif of Programmed Death 1 upon Primary Human T Cell Stimulation, but Only Receptor Ligation Prevents T Cell Activation1.
[15] Xiangmin Li, Zhaoguo Xu, Guoyuan Cui, Li Yu, Xiaoye Zhang.(2020). BTLA Expression in Stage I–III Non–Small-Cell Lung Cancer and Its Correlation with PD-1/PD-L1 and Clinical Outcomes.
[16] Minglei Yang, Chenxi Zheng, Yu Miao, Cuicui Yin, Longfei Tang, Chongli Zhang, Pu Yu, Qingfang Han, Yihui Ma, Shenglei Li, Guozhong Jiang, Wencai Li, Peiyi Xia.(2025). BTLA promoter hypomethylation correlates with enhanced immune cell infiltration, favorable prognosis, and immunotherapy response in melanoma.
[17] Suzanne Mélique, Aurélie Vadel, Nelly Rouquié, Cui Yang, Cyrielle Bories, Coline Cotineau, Abdel Saoudi, N. Fazilleau, Renaud Lesourne.(2024). THEMIS promotes T cell development and maintenance by rising the signaling threshold of the inhibitory receptor BTLA.
[18] Shane Atwell, T. Cheung, Elaine M Conner, Carolyn Ho, Jiawen Huang, Erin L Harryman, Ricky Lieu, Stacie Lim, Wai W Lin, Diana I Ruiz, Andrew C Vendel, Carl F Ware.(2025). Quantitative detection of the HVEM-BTLA checkpoint receptor cis-complex in human lymphocytes.
[19] N. Aubert, S. Brunel, D. Olive, G. Marodon.(2021). Blockade of HVEM for Prostate Cancer Immunotherapy in Humanized Mice.
[20] C. Demerlé, L. Gorvel, M. Mello, S. Pastor, C. Degos, A. Zarubica, F. Angelis, F. Fiore, J. Nunès, B. Malissen, L. Greillier, G. Guittard, H. Luche, F. Barlesi, D. Olive.(2023). Anti-HVEM mAb therapy improves antitumoral immunity both in vitro and in vivo, in a novel transgenic mouse model expressing human HVEM and BTLA molecules challenged with HVEM expressing tumors.
[21] Karolina Wojciechowicz, Katarzyna Kuncewicz, Jacek Rutkowski, Jacek Jassem, Anna Wardowska, M. Spodzieja.(2024). The effect of gD-derived peptides on T cell immune response mediated by BTLA-HVEM protein complex in melanoma patients.
[22] C. Battin, J. Leitner, Petra Waidhofer-Söllner, K. Grabmeier-Pfistershammer, D. Olive, P. Steinberger.(2022). BTLA inhibition has a dominant role in the cis-complex of BTLA and HVEM.
[23] R. Flynn, Tarun E. Hutchinson, K. Murphy, C. Ware, M. Croft, Shahram Salek-Ardakani.(2013). CD8 T Cell Memory to a Viral Pathogen Requires Trans Cosignaling between HVEM and BTLA.
[24] Bin Yang, Zhuochun Huang, Wei-hua Feng, Wei Wei, Junlong Zhang, Y. Liao, Linhui Li, Xinle Liu, Zhiqiang Wu, B. Cai, Yang-juan Bai, Lanlan Wang.(2016). The Expression of LIGHT Was Increased and the Expression of HVEM and BTLA Were Decreased in the T Cells of Patients with Rheumatoid Arthritis.
[25] Weiwei He, Jing Zhao, Xue-rong Liu, Sheli Li, K. Mu, Jing Zhang, Jin-an Zhang.(2020). Associations between CD160 polymorphisms and autoimmune thyroid disease: a case-control study.
[26] Yanbo Kou, Xingping Zheng, Liyuan Meng, Mengnan Liu, Shihong Xu, Qiyue Jing, Shenghan Zhang, Hanying Wang, Jinzhi Han, Zhuanzhuan Liu, Yanxia Wei, Yugang Wang.(2022). The HVEM-BTLA Immune Checkpoint Restrains Murine Chronic Cholestatic Liver Injury by Regulating the Gut Microbiota.
[27] B. Érsek, P. Silló, Uğur Çakır, V. Molnar, A. Bencsik, Balázs Mayer, É. Mezey, S. Kárpáti, Z. Pós, K. Nemeth.(2020). Melanoma-associated fibroblasts impair CD8+ T cell function and modify expression of immune checkpoint regulators via increased arginase activity.
[28] L. Gorvel, C. Demerlé, M. Mello, S. Pastor, C. Degos, A. Zarubica, F. Angelis, Frederic Fiore, Jacques A. Nunès, Bernard Malissen, Laurent Greillier, G. Guittard, Hervé Luche, F. Barlesi, Daniel Olive.(2023). Abstract 5184: Anti-HVEM mAb therapy improves antitumoral immunity both in vitro and in vivo, in a novel transgenic mouse model expressing human HVEM and BTLA molecules challenged with HVEM expressing tumors.
[29] Chong-bin Hu, Chen Huang, Jie Wang, Yun Hong, Dong-dong Fan, Ye Chen, Ai-fu Lin, L. Xiang, J. Shao.(2023). Novel PD-L1/BTLA Checkpoint Axis Exploited for Bacterial Immune Escape by Restraining CD8+ T Cell-Initiated Adaptive Immunity in Zebrafish.
[30] Ryohei Kondo, Kohei Kondo, Kei Nabeshima, Akihiko Nishikimi, Y. Ishida, Toshiaki Shigeoka, Johannes M. Dijkstra.(2025). PD-1 is conserved from sharks to humans: new insights into PD-1, PD-L1, PD-L2, and SHP-2 evolution.
[31]Zhigang Rong, Fei Zhang, Zhengdong Wang, Weifeng He, S. Dong, Jianzhong Xu, F. Dai.(2018). Improved Osteogenesis by HVEM-Expressing Allogenic Bone Marrow-Derived Mesenchymal Stem Cells in an Immune Activation Condition and Mouse Femoral Defect Model.
[32] M. Marasco, Anna Berteotti, J. Weyershaeuser, N. Thorausch, Justyna Sikorska, J. Krausze, H. Brandt, Joanna Kirkpatrick, P. Ríos, W. Schamel, M. Köhn, T. Carlomagno.(2020). Molecular mechanism of SHP2 activation by PD-1 stimulation.
[33] Ling Liu, Yan Cheng, Zhigang Zhang, Jing Li, Y. Geng, Qingsong Li, D. Luo, Li Liang, Wei Liu, Jianping Hu, W. Ouyang.(2023). Study on the allosteric activation mechanism of SHP2 via elastic network models and neural relational inference molecular dynamics simulation.
[34] C. Foster, Jasper Du, Oscar Pundel, M. Geer, Ryan C. Ripert, Jia Liu, Taylor A. Heim, K. Araki, Amanda W. Lund, Jun Wang, Ben Neel.(2025). T lymphocyte-specific deletion of SHP1 and SHP2 promotes activation-induced cell death of CD4+ T cells and impairs antitumor response.
[35] Christine Quach, Xin Li, Pedram Shafiei-Jahani, Meng Li, Stephen Shen, D. Helou, Benjamin P. Hurrell, P. Soroosh, Omid Akbari.(2025). BTLA agonist attenuates Th17-driven inflammation in a mouse model of steroid-resistant asthma.
[36] Jingjing Song, Lihui Wu.(2020). Friend or Foe: Prognostic and Immunotherapy Roles of BTLA in Colorectal Cancer.
[37] P. Diefenhardt, M. Braumann, Thomas Schömig, Bastian Trinsch, Claudio Sierra Gonzalez, J. Becker-Gotot, L. Völker, Lioba Ester, Amrei M. Mandel, D. Hawiger, Ali T. Abdallah, B. Schermer, H. Göbel, P. Brinkkötter, C. Kurts, T. Benzing, Sebastian Brähler.(2023). Stimulation of Immune Checkpoint Molecule B and T-Lymphocyte Attenuator Alleviates Experimental Crescentic Glomerulonephritis.
[38] F. Anzengruber, D. Ignatova, T. Schlaepfer, Yun-Tsan Chang, L. French, S. Pascolo, E. Contassot, M. Bobrowicz, W. Hoetzenecker, E. Guenova.(2019). Divergent LAG-3 versus BTLA, TIGIT, and FCRL3 expression in Sézary syndrome.
[39] A. Ruggieri, M. Yarchoan, S. Goyal, Yuan Liu, E. Sharon, Helen X. Chen, Brian M Olson, C. Paulos, B. El-Rayes, S. Maithel, N. Azad, G. Lesinski.(2022). Combined MEK/PD-L1 inhibition alters peripheral cytokines and lymphocyte populations correlating with improved clinical outcomes in advanced biliary tract cancer.
[40] W. Truong, J. C. Plester, W. Hancock, S. Merani, T. Murphy, K. Murphy, J. Kaye, C. Anderson, A. M. Shapiro.(2007). Combined Coinhibitory and Costimulatory Modulation with Anti‐BTLA and CTLA4Ig Facilitates Tolerance in Murine Islet Allografts.
[41] Radhika R. Gudi, Subha Karumuthil‐Melethil, Nicolas Pérez, Gongbo Li, C. Vasu.(2019). Engineered Dendritic Cell-Directed Concurrent Activation of Multiple T cell Inhibitory Pathways Induces Robust Immune Tolerance.
[42] Tony T. Jiang, O. Kruglov, G. Lin, Angela Minic, Kimberly R. Jordan, R. Uger, M. Wong, Y. Shou, O. Akilov.(2021). Clinical Response to Anti-CD47 Immunotherapy Is Associated with Rapid Reduction of Exhausted Bystander CD4+ BTLA+ T Cells in Tumor Microenvironment of Mycosis Fungoides.
[43] A. Kosmaczewska, L. Ciszak, Anna Andrzejczak, A. Tomkiewicz, Anna Partyka, Zofia Rojek-Gajda, Irena Frydecka, Dariusz Wołowiec, Tomasz Wróbel, A. Bojarska-Junak, Jacek Roliński, Lidia Karabon.(2025). miR-155-5p Silencing Does Not Alter BTLA Molecule Expression in CLL T Cells: Implications for Targeted Immunotherapy.
[44] D. Nishizaki, Sharon Choi, Chinmayi Pandya, Suzanna Lee, S. Pabla, P. DePietro, Taylor J. Jensen, R. Kurzrock, S. Kato.(2025). Pan-Cancer Landscape of B- and T-Lymphocyte Attenuator: Implications for Potential Immunotherapy Combinations.
[45] Binghao Li, Qinchuan Wang, Yihong Luo, Sicong Wang, Sai Pan, Wenting Zhao, Zhaoming Ye, Xifeng Wu.(2024). Peripheral Soluble Immune Checkpoint-Related Proteins Were Associated with Survival and Treatment Efficacy of Osteosarcoma Patients, a Cohort Study.
[46] Patrícia Neuperger, K. Szalontai, Nikolett Gémes, Jozsef A Balog, László Tiszlavicz, J. Furák, György Lázár, László G. Puskás, G. Szebeni.(2023). Single-cell mass cytometric analysis of peripheral immunity and multiplex plasma marker profiling of non-small cell lung cancer patients receiving PD-1 targeting immune checkpoint inhibitors in comparison with platinum-based chemotherapy.
[47] J. Ries, Leah Trumet, Alina Hahn, Lina Kunater, Rainer Lutz, C. Geppert, M. Kesting, Manuel Weber.(2024). The Immune Checkpoint BTLA in Oral Cancer: Expression Analysis and Its Correlation to Other Immune Modulators.
[48]Michelle E Wakeley, Brandon E Armstead, Chyna C. Gray, Elizabeth W. Tindal, Daithi S. Heffernan, C. Chung, A. Ayala.(2023). Lymphocyte HVEM/BTLA co-expression after critical illness demonstrates severity indiscriminate upregulation, impacting critical illness-induced immunosuppression.
[49]Julie Joseph, T. Premeaux, Daniel O. Pinto, Ahbishek Rao, Shrobona Guha, A. Panfil, A. Carey, L. Ndhlovu, E. Bergmann-Leitner, P. Jain.(2023). Extracellular immune checkpoint molecules released from HTLV-1-infected cells mount immune suppression in the context of neuroinflammation.
[50] Chien-Hsun Huang, Lei Cong, Jun Xie, Bo Qiao, S. Lo, T. Zheng.(2009). Rheumatoid arthritis-associated gene-gene interaction network for rheumatoid arthritis candidate genes.
[51] A. Small, S. Cole, J. J. Wang, S. Nagpal, Ling-Yang Hao, M. Wechalekar.(2022). Attenuation of the BTLA/HVEM Regulatory Network in the Circulation in Primary Sjögren’s Syndrome.
[52] Haruka Kinosada, Jun-ichirou Yasunaga, Kazuya Shimura, P. Miyazato, Chiho Onishi, T. Iyoda, K. Inaba, M. Matsuoka.(2017). HTLV-1 bZIP Factor Enhances T-Cell Proliferation by Impeding the Suppressive Signaling of Co-inhibitory Receptors.
[53] Kai Werner, S. Dolff, Yang Dai, Xin Ma, A. Brinkhoff, J. Korth, A. Gäckler, H. Rohn, Ming Sun, J. C. Cohen Tervaert, P. van Paassen, A. Kribben, O. Witzke, B. Wilde.(2019). The Co-inhibitor BTLA Is Functional in ANCA-Associated Vasculitis and Suppresses Th17 Cells.
[54] Bin Yang, Junlong Zhang, Lixin Li, Xiaojun Lyu, Wei Wei, Zhuochun Huang, B. Cai, Lanlan Wang.(2017). Genetic variations in LIGHT are associated with susceptibility to ankylosing spondylitis in a Chinese Han population.
[55] H. Douna, J. Amersfoort, F. Schaftenaar, M. Kröner, Máté G Kiss, B. Slütter, M. Depuydt, Mireia N A Bernabé Kleijn, A. Wezel, H. Smeets, H. Yagita, C. Binder, I. Bot, G. V. van Puijvelde, J. Kuiper, A. Foks.(2019). BTLA stimulation protects against atherosclerosis by regulating follicular B cells.
[56] P. Diefenhardt, Marie Braumann, Bastian Trinsch, Thomas Schömig, Claudio Sierra Gonzalez, Bernhard Schermer, Thomas Benzing, P. Brinkkoetter, Sebastian Braehler.(2022). Immune Checkpoint Molecule BTLA Attenuates Experimental Glomerulonephritis by Directly Inhibiting T Effector Cells and Inducing Treg Differentiation.


Comments
Leave a Comment