Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Coagulation, also known as clotting, is the process by which blood changes from a liquid to a gel and form a blood clot. The formation of a clot is often referred to as secondary hemostasis, because it forms the second stage in the process of arresting the loss of blood from a ruptured vessel. The first stage, primary hemostasis, is characterized by blood vessel constriction and platelet aggregation at the site of vessel injury. The mechanism of coagulation involves activation, adhesion, and aggregation of platelets along with deposition and maturation of fibrin. Disorders of coagulation are disease states which can result in bleeding or obstructive clotting (thrombosis).
Coagulation is the biological process by which blood transforms into solid form. The critical function of coagulation mainly includes two points. One is to seal any breach of a human body’s primary organ of protection against harmful, external agents such as germs - the skin, and another is to maintain the closed circuit integrity necessary for the body to deliver oxygen and nutrients to all of its tissues via the circulatory system. As well as the process is common to many nonhuman animals, and can be thought of as nature's version of a bandage.
Clotting is a sequential process that involves the interaction of numerous blood components called coagulation factors. There are 12 principal coagulation factors in all, and each of these has been assigned a roman numeral, I to XIII. As shown in Table 1, maybe you will confuse where is the number VI? Number VI, also known as Factor Va, has been unassigned, because it actually is the form of activated V factor. Note that, coagulation factor I, also known as fibrinogen, the final secreted, hepatocyte-derived glycoprotein is composed of two trimers with each trimer composed of three different polypeptide chains, including fibrinogen alpha chain (α chain) (encoded by the FGA gene), fibrinogen beta chain (β chain) (encoded by the FGB gene), and fibrinogen gamma chain (the γ chain) (encoded by the FGG gene). Besides that, coagulation factor XIII is a 320kDa glycoprotein tetramer consisting of twice two subunits (2 A and 2 B).
Table 1. the family of coagulation factors
| Number | Name/ Alias | Chemical Essence | Synthetic site | Involving pathway | Function |
|---|---|---|---|---|---|
| I | Fibrinogen | glycoprotein | Liver | Final common pathway | Forms clot (fibrin) |
| II | Prothrombin | glycoprotein | Liver | Final common pathway | Its active form (IIa) activates I, V, X, VII, VIII, XI, XIII, protein C, platelets |
| III | Tissue factor | glycoprotein |
Tissue Endotheliocyte monocyte |
Tissue factor pathway | Co-factor of VIIa (formerly known as factor III) |
| IV | Calcium | calcium ion | - | Three pathways | Required for coagulation factors to bind to phospholipid (formerly known as factor IV) |
| V | proaccelerin, labile factor | glycoprotein | Liver | Final common pathway | Co-factor of X with which it forms the prothrombinase complex |
| VII | stable factor, proconvertin | glycoprotein | Liver | Tissue factor pathway | Activates IX, X |
| VIII | Antihemophilic factor A | glycoprotein | Liver | Contact activation pathway | Co-factor of IX with which it forms the tenase complex |
| IX | Antihemophilic factor B | glycoprotein | Liver | Contact activation pathway | Activates X: forms tenase complex with factor VIII |
| X | Stuart-Prower factor | glycoprotein | Liver | Final common pathway | Activates II: forms prothrombinase complex with factor V |
| XI | plasma thromboplastin antecedent | glycoprotein | Liver | Contact activation pathway | Activates IX |
| XII | Hageman factor | glycoprotein | Liver | Contact activation pathway | Activates factor XI, VII and prekallikrein |
| XIII | fibrin-stabilizing factor | glycoprotein | Liver, platelet | Final common pathway | Crosslinks fibrin |
*the content of Table 1 refers to the wiki
Cascade, as its name means, is a series of steps or stages (as of a physiological process) which, once initiated, continues to the final step because each step is triggered by the preceding one, resulting in amplification of the signal, information, or effect at each stage. A coagulation cascade is the series of steps beginning with activation of the contact activation pathway of coagulation or tissue factor pathways of coagulation, or of one of the related alternative pathways, and proceeding through the common pathway of coagulation to the formation of the fibrin clot. The contact activation pathway of coagulation forms clots in response to abnormalities in the wall of a blood vessel in the absence of tissue injury. And tissue factor pathway activates clotting as a result of tissue injury. The two pathways start differently and then both progress to the final common pathway. Both pathways depend on multiple clotting factors (shown in Table 2). These clotting factors are present in the body in inactive forms until they are activated during the coagulation cascade. The connection among of three pathway is shown in the Fig. 1.
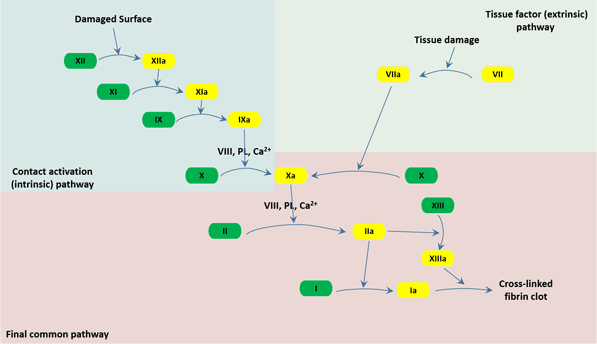
Fig.1. The three pathways that makeup the classical blood coagulation pathway
Tissue factor pathway, also known as extrinsic pathway, is the most frequent and most familiar of the two pathways of the coagulation cascade. When a tissue damage such as a cut or a contusion causes bleeding, VII leaves the circulation combines with a glycoprotein, which called tissue factor (TF), expressed on tissue-factor-bearing cells to form an activated complex (TF-VIIa). Then TF-VIIa activates IX and X. Next, factor VIIa then cleaves factor X molecules circulating in the blood to form factor Xa. From this point on, the tissue factor and contact activation pathways are the same and are known as the combined pathway[1].
The contact activation of the coagulation cascade begins when blood comes into contact with the collagen in the damaged wall of a blood vessel and forms the primary complex on collagen by high-molecular-weight kininogen (HMWK), prekallikrein, and XII. This causes factor XII, the Hageman factor, converting into its factor XIIa. Factor XIIa actives factor XI into factor XIa, which converts factor IX into factor IXa. Hemophilia B is a genetic bleeding disorder characterized by a deficiency in factor IX. Factor IXa converts factor X into factor Xa; this is the point at which the intrinsic pathway joins the combined pathway[2].
In the final common pathway, also called the combined pathway, factor Xa activates factor II to form factor IIa (thrombin). Thrombin activates the blood's platelets and leads them to gather together at the affected site and form a platelet plug. A substance, which named von Willebrand factor (vWF), makes it possible for platelets to stick to the injured site. A deficiency of vWF causes von Willebrand disease, the most common inherited bleeding disorder.
Coagulation defects may cause hemorrhage or thrombosis, and occasionally both, depending on the nature of the defect. In this section, we exemplify several diseases caused by the coagulation disorders, including coagulation disorders, platelet disorders, inflammation and some other special diseases.
Coagulation disorders are disruptions in the body's ability to control blood clotting. The most commonly known coagulation disorder is hemophilia (PWH), a condition in which a critical component of blood coagulation is missing, causing individuals to bleed for long periods of time before clotting occurs. There are numerous other coagulation disorders stemming from a variety of causes. Emerging evidence revealed that a diversity of coagulation disorders in cancer patients arise from tumor-specific growth characteristics, defective myelopoiesis, hypoproteinemia, neoangiogenesis with impaired endothelial lining, or metastatic lesions growth with organ dysfunction[3]. Besides that, recent investigations have discovered a clinically relevant correlation of coagulation disorders and tumor growth. About treatment in persons with hemophilia, the current standard treatment is prophylaxis, given intravenously twice or thrice weekly, which is associated with a non-negligible burden on patients' quality of life[4]. Furthermore, Swystun LL's team have found a gene therapy for coagulation disorders[5].
Platelet disorders, also called thrombocyte disorders, mainly involves three types of disorders, including an abnormal increase in platelets (thrombocythemia and reactive thrombocytosis), a decrease in platelets (thrombocytopenia) and platelet dysfunction. Platelets are little pieces of blood cells. The primary function of platelet is to help wounds heal and prevent bleeding by forming blood clots. And the platelet derives from bone marrow. If your blood has a low concentration of platelets, you may have a higher risk of mild to serious bleeding. While if your blood has too many platelets, it may increase your risk of blood clots. Besides that, platelet dysfunction means that the platelet do not work as they should. For instance, in von Willebrand Disease, the platelets cannot stick together or attach to blood vessel walls well so that excessive bleeding. Recently, accumulating evidence have demonstrated that inherited platelet disorders can be diagnosed rapidly by high-throughput sequencing[6][7][8].
Over 150 years ago, Anatomic pathology studies revealed that excessive activation of coagulation occurs in the setting of inflammation[9]. But the molecular mechanisms by which these systems interact doesn't delineate until recent decades. Although viewed separately, coagulation and inflammation are highly integrated, delicately balanced biological systems with extensive cross talk that optimizes the organism's response to injury and invasion by pathogens. In this section, we highlight some key molecular interactions between them, some of which are already showing promise as therapeutic targets for inflammatory and thrombotic disorders.
During injury or infection, circulating monocytes (M) and subendothelial cells (smooth muscle cells, SMC, et al.) are activated via reactive oxygen species (ROS), cytokines or endotoxin (LPS), and damage-associated molecular patterns (DAMPs) though pathogen recognition receptors (eg. Toll-like receptors (TLRs)). TF is exposed to the blood, sustained through the release of pro-inflammatory cytokines, chemokines, and inflammatory by leukocytes, platelets, endothelial cells, and perivascular cells. TF triggers the series of factor VIIa, factor Xa, and thrombin (IIa), as shown in Fig.1, which activate platelets and induce protease-activated receptor (PAR)–mediated signaling, with release of more inflammatory cytokines, upregulation of leukocyte adhesion molecules (intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1)), and suppression of vasculoprotective molecules, such as thrombomodulin (TM). Release of P-selectin (CD62), von Willebrand Factor (VWF), platelet factor 4 (PF4), and CD40 ligand overall promotes recruitment, adhesion, and activation of platelets and leukocytes with formation of neutrophil extracellular traps (NETs). C5a and C5b-9 may be generated after exposure to pathogens and damaged host cells, facilitated by thrombin. These products further recruit inflammatory leukocytes and promote clotting and cytokine release[10][11].
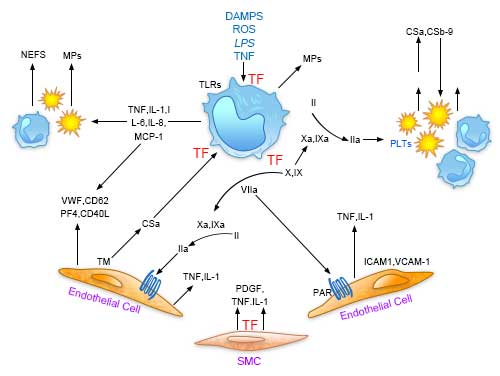
Fig. 2. Central role of tissue factor (TF) in the interplay between coagulation and inflammation.
On contact with highly anionic surfaces (not shown), factor XII (FXII) autoactivates to generate small amounts of FXIIa. FXIIa recruits high molecular weight kininogen (HK/HKa) that is complexed with prekallikrein (PPK). FXIIa activates PPK to kallikrein (PK), which in turn generate more FXIIa and activates HK to release bradykinin (BK). BK binds to its receptors (B2R) on endothelial cells and neutrophils, triggering proinflammatory responses. FXIIa also activates FXI in the coagulation pathway, inducing further thrombin (IIa) generation. When free, IIa induces coagulation and inflammation. When bound to thrombomodulin (not shown), IIa promotes generation of activated protein C (APC) and TAFIa. Not shown are activating surfaces and C1-inhibitor, the major negative regulator of FXIIa, FXIa, and PK. TAFI indicates thrombin activatable fbrinolysis inhibitor[12[13].
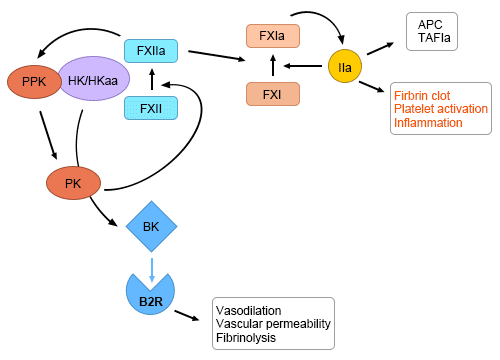
Fig.3. Factor XII activation at the nexus of coagulation and inflammation.
Besides that, if you want to obtain the more information about interactions, please click here to view the details.
Coagulation is a dynamic process and the understanding of the blood coagulation system has evolved over the recent years in anaesthetic practice. In the rest section of this article, we introduce two related clinical applications of coagulation.
Coagulation tests are similar to most blood tests. Side effects and risks are minimal. A medical professional will take a blood sample and send it to a laboratory for testing and analysis. Conditions that can cause coagulation problems include liver disease, thrombophilia, which is excessive clotting, and hemophilia, which is an inability to clot normally. Coagulation tests are useful in monitoring people who take medications that affect clotting ability. Coagulation tests are also sometimes recommended before surgery. There are many types of coagulation tests, including explanations of several of them, including complete blood count (CBC), factor V assay, fibrinogen level, prothrombin time (PT or PT-INR), platelet count, thrombin time and bleeding time.
Coagulation is a tightly controlled biochemical network of reactions that leads to the generation of thrombin [14]. This network includes zymogens, enzymes, co-factors, and inhibitors, which quickly generate thrombin at the site of injury and efficiently eliminate it in the peripheral area. Thrombin-antithrombin complex (TAT) is a small protein molecule complex, several enzymes of the coagulation system can be inactivated. Thrombin-antithrombin complex is a glycoprotein produced by the liver, the 432 amino acids.
Thrombin-antithrombin complex in plasma half-life of about three days. In normal plasma thrombin-antithrombin complex concentration of about 0.12 mg / ml. From the thrombin-antithrombin complex has been a large number of other species fractionated protein and cDNA sequencing showed that the amino acid residue in the amino acid residue at the sixth position locus. Three single body of six amino acids form a disulfide bond: Cys8-Cys128, Cys21-Cys95, Cys248-Cys430. Four potential N- glycosylation sites are located in the primary structure of thrombin-antithrombin complex position 96,135,155 and 192 asparagine (Asn) residues in other species of anti- thrombin also occurs in a similar position. In the main form of thrombin-antithrombin complex, all of these loci are stuck oligosaccharide side chains covalently attached, resulting in this form of the molecular weight of up to 58,200 anti-thrombin. Secondary situation Thrombin-antithrombin complex, the potential glycosylation site at asparagine 135 is not occupied.
Thrombin-antithrombin complex elisa kit uses a two-step double-antibody sandwich enzyme-linked immunosorbent assay (ELISA). The standard, pre-coated test samples was added to TAT monoclonal antibody coated microtiter plate transparent, sufficient incubation time, washed to remove unbound components, adding enzyme working solution , sufficient incubation time, washed to remove unbound components. Sequentially adding the substrate A, B, substrate (TMB) horseradish peroxidase (HRP) catalyzed conversion of the blue product, under the action of an acid becomes yellow, the color depth of the sample of human anti-clotting enzyme TAT are positively correlated, 450nm wavelength measured OD values, according to the standards and samples OD values, calculate the sample of TAT content.
References:
[1] Pallister CJ, Watson MS. Haematology [M]. Scion Publishing. 2010, 336–347.
[2] Long, Andrew T.; Kenne, Ellinor;et al. Contact system revisited: An interface between inflammation, coagulation, and innate immunity [J]. Journal of Thrombosis and Haemostasis. 2015, 14: 427–437.
[3] Kvolik S, Jukic M, et al. An overview of coagulation disorders in cancer patients [J]. Surg Oncol. 2010, 19: 33-46.
[4] Mannucci PM, Mancuso ME. Investigational drugs for coagulation disorders [J]. Expert Opin Investig Drugs. 2013, 22(8): 945-53.
[5] Swystun LL, Lillicrap D. Gene Therapy for Coagulation Disorders [J]. Circ Res. 2016, 118(9):1443-52.
[6] Nurden AT, Nurden P. High-throughput sequencing for rapid diagnosis of inherited platelet disorders: a case for a European consensus [J]. Haematologica. 2018, Jan, 103(1):6-8.
[7] Bastida JM, Lozano ML, et al. Introducing high-throughput sequencing into mainstream of genetic diagnosis practice in inherited platelet disorders [J]. Haematologica, 2017.
[8] Freson K, Turro E. High-throughput sequencing approaches for diagnosing hereditary bleeding and platelet disorders [J]. J Thromb Haemost. 2017, 15(7): 1262-1272.
[9] Carmeliet P, Mackman N, et al. Role of tissue factor in embryonic blood vessel development [J]. Nature. 1996. 383:73–75.
[10] Jonathan H. Foley, Edward M. Conway. Cross Talk Pathways Between Coagulation and Inflammation [J]. Circ Res. 2016, 118(9):1392-408.
[11] Bogdanov VY, Balasubramanian V, eet al. Alternatively spliced human tissue factor: a circulating, soluble, thrombogenic protein [J]. Nat Med. 2003, 9:458–462.
[12] Oschatz C, Maas C, et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo [J]. Immunity. 2011, 34:258–268.
[13] Vague P, Juhan-Vague I, et al. Correlation between blood fbrinolytic activity, plasminogen activator inhibitor level, plasma insulin level, and relative body weight in normal and obese subjects. Metabolism. 1986, 35:250–253.
[14] Kuldeepsinh Rana a, Keith B. Neeves. Blood flow and mass transfer regulation of coagulation [J]. Blood Rev.2016 30(5):357-68.


Comments
Leave a Comment