Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Apoptosis (programmed cell death) and autophagy (lysosome-mediated self-degradation) are not independent cell fate pathways. Their dynamic, bidirectional crosstalk dictates cancer cell survival and response to therapy. This article reviews the shared molecular regulators that govern apoptosis-autophagy interaction, explains how cancer cells hijack this network to develop resistance, and highlights actionable therapeutic strategies targeting their intersection. We conclude that combinatorial approaches modulating both pathways hold greater promise than single-pathway therapies for improving cancer outcomes [1].
Table of Contents
1. Why Study Apoptosis-Autophagy Crosstalk in Cancer?
2. What Do We Need to Know About Apoptosis and Autophagy?
3. Where Do Apoptosis and Autophagy Intersect at the Molecular Level?
4. How Does Apoptosis-Autophagy Crosstalk Shape Cancer Biology?
5. Target Apoptosis-Autophagy Crosstalk for Better Cancer Therapy
6. Challenges and Future Directions of Apoptosis-Autophagy Crosstalk Research
Apoptosis and autophagy are two tightly regulated programs that decide whether a cell survives, adapts, or dies. In cancer, these systems rarely act in isolation; instead, they are linked by shared signaling nodes and can either cooperate or oppose each other depending on context [2].
Understanding how apoptosis and autophagy intersect is central to explaining why tumors arise, how they progress, and why they respond or resist therapy. This crosstalk also creates both opportunities and traps when we try to target these pathways with drugs.
Before diving into their interaction, we briefly review the core concepts of each pathway, focusing only on information critical for understanding crosstalk.
Apoptosis is a highly regulated form of cell death that eliminates damaged or unnecessary cells without causing inflammation. It proceeds through two primary pathways:
Autophagy is an evolutionarily conserved process that degrades and recycles damaged organelles and misfolded proteins. The autophagy process involves three key steps:
AMP-activated protein kinase (AMPK), the cell's energy sensor, is the primary positive regulator of autophagy, activating it when cellular energy levels are low [2,4].
Autophagy inhibits apoptosis under sublethal stress to promote cell survival. During nutrient/energy deprivation, autophagy catabolizes cellular components to supply nutrients and ATP, sustaining cell viability and blocking apoptosis. Genetic ablation of core autophagy genes (Atg5, Atg7) causes lethal apoptosis in neonatal mice during the post-birth starvation period [25].
Under endoplasmic reticulum (ER) stress, autophagy degrades unfolded protein aggregates to alleviate ER stress, thereby inhibiting ER stress-mediated apoptotic signaling [26].
Apoptosis reciprocally inhibits autophagy during its execution phase. Activated caspase-3 cleaves the essential autophagy mediator Beclin 1, inactivating Beclin 1-dependent autophagic flux. This cleavage terminates pro-survival autophagy and generates a truncated Beclin 1 fragment that further amplifies apoptotic signaling, forming a feedforward loop to lock in cell death [27].
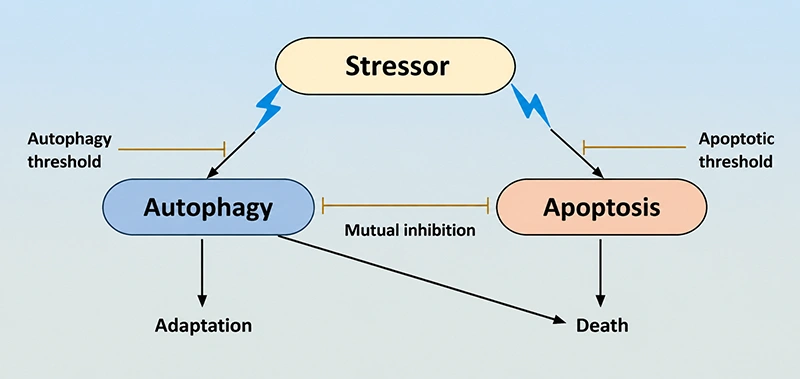
Figure 1. The relationship between apoptosis and autophagy
Autophagy primarily acts as a pro-survival mechanism by degrading damaged organelles and misfolded proteins for metabolic recycling, while apoptosis executes programmed cell death to eliminate compromised or unwanted cells. Despite their distinct functional endpoints, these two pathways are deeply interconnected via a network of shared molecular regulators, known as crosstalk factors. These factors dynamically modulate the balance between cell survival and death in a context- and stimulus-dependent manner.
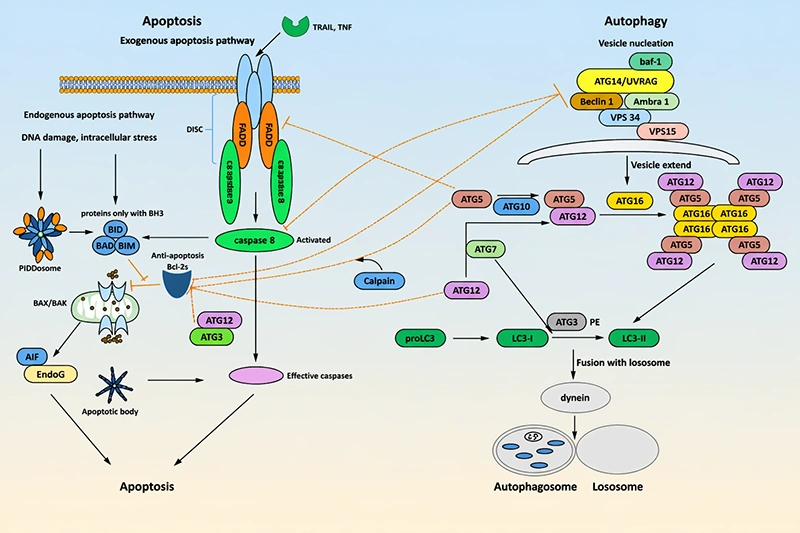
Figure 2. The key crosstalk factors of apoptosis and autophagy
The Bcl-2 protein family is the most well-characterized molecular bridge between autophagy and apoptosis, serving as a master regulator of the cell survival-death balance [5,6]. This family is classified into three subclasses: anti-apoptotic proteins (Bcl-2, Bcl-xL, Mcl-1, Bcl-w), pro-apoptotic effector proteins (Bax, Bak), and pro-apoptotic BH3-only proteins (Bad, PUMA, Noxa, Bim, Bid). Bioactive sphingolipids, such as ceramide and sphingosine-1-phosphate, also modulate Bcl-2 family activity to coordinate autophagy and apoptosis signaling [7].
In the apoptotic pathway, anti-apoptotic Bcl-2 proteins sequester Bax/Bak to prevent MOMP, the irreversible commitment step of intrinsic apoptosis. BH3-only proteins, activated by stress signals, displace anti-apoptotic Bcl-2 proteins from Bax/Bak, enabling Bax/Bak oligomerization, MOMP, cytochrome c release, and apoptosome formation with subsequent caspase activation [5,6].
In parallel, Bcl-2 family proteins directly regulate autophagy via interaction with Beclin-1, the core initiator of autophagosome nucleation. Beclin-1 contains a BH3 domain that mediates its direct binding to the anti-apoptotic Bcl-2, Bcl-xL, and Mcl-1. This binding sequesters Beclin-1 from the class III PI3KC3 complex (comprising Vps34, Vps15, Atg14), thereby inhibiting autophagy initiation [5,6].
Under metabolic stress, BH3-only proteins (e.g., Bad, BNIP3) or BH3 mimetics disrupt the Bcl-2-Beclin-1 interaction, releasing Beclin-1 to assemble the PI3KC3 complex and trigger autophagy. Notably, post-translational modifications of Bcl-2, such as phosphorylation at Ser70 by AMPK, also dissociate Bcl-2 from Beclin-1 to promote autophagy, while dephosphorylation stabilizes the inhibitory complex [5].
This dual regulatory role of Bcl-2 family proteins creates a direct molecular link: the same pool of Bcl-2 proteins simultaneously modulates the threshold for both apoptosis and autophagy, enabling coordinated cellular responses to stress. Dysregulation of this axis is frequently observed in cancer, where Bcl-2 overexpression simultaneously suppresses apoptosis and autophagy, promoting tumor survival and chemoresistance [8][9].
Caspases, a family of cysteine-dependent aspartate-directed proteases, are the core executioners of apoptosis, and also act as critical molecular switches that terminate autophagy and drive the transition to apoptotic cell death [10]. Caspases are categorized into initiator caspases (caspase-2, caspase-8, caspase-9, caspase-10) and effector caspases (caspase-3, caspase-6, caspase-7), all of which are synthesized as inactive zymogens and activated via proteolytic cleavage during apoptosis.
Activated caspases directly inhibit autophagy by cleaving and inactivating key autophagy-related (ATG) proteins. The most well-documented substrate is Beclin-1: caspase-3, -7, and -8 cleave Beclin-1 at multiple aspartate residues, generating C-terminal and N-terminal fragments that lose their autophagy-inducing activity. Notably, the C-terminal cleavage fragment of Beclin-1 translocates to mitochondria and amplifies apoptotic signaling by promoting cytochrome c release, creating a positive feedback loop that reinforces the switch from autophagy to apoptosis [11,12].
Caspases also target other core ATG proteins. Atg5, a key component of the Atg5-Atg12-Atg16L complex required for autophagosome elongation, is cleaved by calpain and caspase-3 to generate a truncated 24 kDa fragment. This fragment loses autophagic activity but gains pro-apoptotic function: it translocates to mitochondria, binds to Bcl-xL, and triggers cytochrome c release and caspase activation [5]. Similarly, Atg7, Atg4D, and Atg16L1 are also caspase substrates, whose cleavage inactivates autophagy and in some cases generates pro-apoptotic fragments [10][12].
Conversely, autophagy can modulate caspase activity in a bidirectional manner. In pro-survival contexts, autophagy sequesters active caspase-8 and caspase-3 into autophagosomes for lysosomal degradation, thereby suppressing apoptosis. However, in certain contexts, autophagosomal membranes serve as a platform for the assembly of the death-inducing signaling complex (DISC), promoting caspase-8 activation and apoptosis initiation [10][12]. This dual role highlights the context-dependent nature of caspase-mediated crosstalk.
Beyond being caspase substrates, several ATG proteins directly participate in apoptotic regulation, independent of their canonical autophagic functions, establishing another layer of crosstalk [11,12].
Atg12, a core component of the autophagosome elongation system, has a BH3-like domain that mediates direct binding to anti-apoptotic Bcl-2 and Mcl-1, neutralizing their anti-apoptotic activity and promoting Bax/Bak-dependent apoptosis. This function is independent of its conjugation to Atg5, demonstrating a non-canonical pro-apoptotic role of Atg12 [12].
Atg7, the E1-like enzyme essential for both Atg12-Atg5 and LC3-PE conjugation, interacts with caspase-9 to modulate intrinsic apoptosis. Under genotoxic stress, Atg7 promotes caspase-9 activation and apoptosis, independent of its autophagic enzymatic activity. Conversely, Atg7-mediated LC3 lipidation is required for the autophagic degradation of active caspases, creating a negative feedback loop on apoptosis [10][12].
The LC3/Atg8 family proteins, canonical markers of autophagosome formation, also directly regulate apoptotic signaling. LC3 interacts with the adaptor protein p62/SQSTM1 to recruit caspase-8 to autophagosomal membranes, promoting its dimerization and activation in certain contexts, such as death receptor stimulation. Additionally, LC3 mediates the selective autophagic degradation of the pro-apoptotic protein PUMA, thereby suppressing apoptosis under nutrient stress [10][12].
The mTOR and AMPK, the two primary cellular energy and nutrient sensors, act as upstream master regulators that coordinately control autophagy and apoptosis, integrating extracellular and intracellular stress signals [9][13].
mTOR, specifically mTOR complex 1 (mTORC1), is a serine/threonine kinase that is activated under nutrient-replete, growth factor-replete conditions. Active mTORC1 directly phosphorylates the ULK1/Atg13/FIP200 complex, inhibiting ULK1 kinase activity and blocking autophagy initiation [9]. In parallel, mTORC1 signaling promotes cell survival and inhibits apoptosis by phosphorylating and inactivating pro-apoptotic proteins such as Bad and upregulating anti-apoptotic Bcl-2 family proteins. Conversely, mTORC1 inhibition (e.g., by nutrient starvation, rapamycin) activates ULK1 to trigger autophagy, while also sensitizing cells to apoptosis via downregulation of anti-apoptotic proteins [9][14].
AMPK, the cellular energy sensor activated by low ATP/AMP ratios (e.g., glucose deprivation, metabolic stress), is a potent activator of autophagy and a dual regulator of apoptosis. AMPK directly phosphorylates and activates ULK1 to initiate autophagy, and also inhibits mTORC1 via phosphorylation of TSC2 and Raptor, reinforcing autophagy induction [14]. In terms of apoptosis regulation, AMPK exerts context-dependent effects: in mild stress, AMPK-mediated autophagy activation suppresses apoptosis to promote cell survival; in severe, sustained stress, AMPK promotes apoptosis by phosphorylating and stabilizing p53, and upregulating pro-apoptotic BH3-only proteins [13][15].
This mTOR-AMPK axis thus acts as a central rheostat that senses cellular metabolic status and coordinates the balance between autophagic survival and apoptotic death [9][14].
p53, the "guardian of the genome", is a critical transcription factor that responds to DNA damage, oncogenic stress, and oxidative stress, and is a key mediator of autophagy-apoptosis crosstalk via dual, compartment-specific functions [9][15].
In the nucleus, activated p53 transcriptionally upregulates a panel of target genes that modulate both autophagy and apoptosis. For apoptosis, p53 induces the expression of pro-apoptotic proteins including Bax, Bak, PUMA, Noxa, and Fas, while downregulating anti-apoptotic Bcl-2 and Bcl-xL, strongly promoting apoptotic cell death in response to irreparable DNA damage [11][15]. For autophagy, nuclear p53 transcriptionally activates AMPK and inhibits mTORC1 via upregulation of TSC2, PTEN, and sestrins, thereby promoting autophagy initiation. Additionally, p53 upregulates the expression of damage-regulated autophagy modulator (DRAM), a lysosomal protein that is required for p53-induced autophagy and can also promote apoptosis in a context-dependent manner [9][15].
In contrast, cytosolic p53 exerts an inhibitory effect on autophagy. Cytosolic p53 directly binds to FIP200, a component of the ULK1 complex, blocking ULK1 activation and autophagy initiation [15]. This dual role creates a dynamic regulatory loop: in response to mild stress, transient p53 activation promotes autophagy to repair damaged DNA and promote cell survival; in response to severe, irreparable damage, sustained nuclear p53 activation drives the switch from autophagy to apoptotic cell death via upregulation of pro-apoptotic target genes [11][15].
Reactive oxygen species (ROS) and endoplasmic reticulum (ER) stress are common upstream triggers of both autophagy and apoptosis, and their downstream mediators act as important crosstalk factors [13][16].
ROS, generated primarily from damaged mitochondria under stress, activates both autophagy and apoptosis in a dose-dependent manner. Low to moderate levels of ROS induce autophagy by inactivating the cysteine protease Atg4 (to promote LC3 lipidation), activating AMPK, and inhibiting mTORC1, thereby promoting mitochondrial quality control (mitophagy) to reduce ROS production and suppress apoptosis. However, high levels of ROS trigger MOMP, cytochrome c release, and caspase activation, while also inactivating anti-apoptotic Bcl-2 proteins, driving apoptotic cell death. Concurrently, excessive ROS promotes the caspase-mediated cleavage of Beclin-1, terminating autophagy and amplifying apoptosis [10][13][16].
ER stress, triggered by the accumulation of unfolded/misfolded proteins in the ER lumen, activates the unfolded protein response (UPR), which consists of three branches: PERK, IRE1α, and ATF6. The UPR initially promotes adaptive autophagy to clear misfolded proteins and resolve ER stress, thereby suppressing apoptosis. PERK activation upregulates ATF4, which transcriptionally induces the expression of Atg5, Atg7, and Beclin-1 to promote autophagy. However, unresolved, chronic ER stress switches the UPR from pro-survival to pro-apoptotic signaling: PERK-ATF4 upregulates the pro-apoptotic transcription factor CHOP, which downregulates anti-apoptotic Bcl-2 and upregulates BH3-only proteins, triggering intrinsic apoptosis. IRE1α activation also promotes apoptosis via activation of JNK and caspase-12, while simultaneously inhibiting autophagy via caspase-mediated cleavage of Beclin-1 [11][13][16].
Cancer cells exploit apoptosis-autophagy crosstalk to survive in hostile environments and resist therapy. Understanding how this occurs is essential for developing effective treatments.
Autophagy exhibits a context-dependent dual role in cancer, which is the primary reason targeting it has been challenging.
In normal tissues, basal autophagy removes damaged mitochondria and misfolded proteins, reducing oxidative stress and genome instability, which are classic drivers of tumorigenesis.
In early tumorigenesis, autophagy acts as a tumor suppressor. It removes damaged organelles and reduces reactive oxygen species (ROS) levels, preventing genomic instability that can lead to malignant transformation [17]. Beclin 1 haploinsufficiency, which impairs autophagy, is found in 40–75% of human breast, ovarian, and prostate cancers [18].
Once tumors are established, the microenvironment becomes hostile, with hypoxia, low nutrients, and therapeutic insults. Under these stresses, autophagy is often activated in poorly vascularized tumor regions and in apoptosis‑defective cells. It provides cancer cells with nutrients and energy to survive metabolic stress, hypoxia, and chemotherapy. Many aggressive cancers become "autophagy-dependent," relying on this process for growth and survival [17].
The most common mechanism of therapy resistance is the induction of cytoprotective autophagy in response to apoptosis-inducing treatments. Chemotherapy, targeted therapy, and immunotherapy all trigger autophagy as a survival response in cancer cells [18].
Cancer cells achieve this through dysregulation of crosstalk molecules:
The expression levels of autophagy and apoptosis markers have significant prognostic value across different cancer types. High LC3-II (a marker of autophagosome formation) expression is generally associated with poor prognosis in established tumors, as it indicates increased autophagic activity supporting cancer cell survival [21].
However, current biomarkers have limitations. They measure static levels of autophagy proteins rather than dynamic autophagic flux, which is the functionally relevant parameter. This has complicated the interpretation of clinical trial results [18].
Targeting apoptosis-autophagy crosstalk offers a promising approach to overcome therapy resistance. Three main therapeutic strategies are currently being investigated in preclinical and clinical studies.
The most widely studied approach is to inhibit cytoprotective autophagy, thereby unleashing the full apoptotic potential of cancer therapies.
Chloroquine (CQ) and hydroxychloroquine (HCQ) are the only autophagy inhibitors currently in late-stage clinical trials. They work by inhibiting lysosomal acidification, preventing autophagosome degradation. Numerous phase I/II trials have shown that combining HCQ with chemotherapy, targeted agents (e.g., EGFR inhibitors), or immune checkpoint inhibitors improves treatment efficacy [18].
Newer, more specific autophagy inhibitors are in development. These include ULK1 inhibitors (e.g., DCC-3116, currently in phase 1 trials) and PPT1 inhibitors (e.g., GNS561), which show improved potency and specificity compared to CQ/HCQ [21]. A phase 2 study evaluating GNS561 in combination with atezolizumab and bevacizumab for unresectable hepatocellular carcinoma is currently recruiting patients [19].
In cancers that have lost apoptotic machinery (e.g., Bax/Bak-deficient tumors), inducing excessive autophagy can trigger autophagic cell death.
mTOR inhibitors (rapamycin and its analogs) are the most commonly used autophagy inducers. They work by inhibiting mTORC1, the central negative regulator of autophagy. AMPK activators such as metformin also induce autophagy and have shown anticancer activity in preclinical studies [20].
However, this approach faces significant challenges. Achieving sufficient autophagy induction to cause cell death is difficult, and autophagy inducers often have off-target effects. Additionally, in most cases, autophagy induction promotes rather than inhibits cancer cell survival [18].
Targeting molecules that regulate both apoptosis and autophagy simultaneously is an emerging and promising strategy.
BH3 mimetics, such as venetoclax (a Bcl-2 inhibitor approved for chronic lymphocytic leukemia), disrupt the interaction between Bcl-2 and both pro-apoptotic Bax/Bak and pro-autophagic Beclin 1. This simultaneously induces apoptosis and modulates autophagy, leading to synergistic cell death [22]. Novel BH3 mimetics that target multiple Bcl-2 family members are showing improved efficacy in solid tumors [24].
p53 modulators that restore wild-type p53 function are also being investigated. These agents reactivate both the apoptotic and autophagic tumor-suppressor functions of p53, thereby making them particularly effective against p53-mutant cancers [23].
Despite significant progress, several key challenges remain in translating apoptosis-autophagy crosstalk research into clinical practice.
The crosstalk between apoptosis and autophagy is a complex, highly regulated network governed by a set of shared molecular factors, including Bcl-2 family proteins/Beclin-1, caspases, ATG proteins, the mTOR-AMPK axis, p53, and ROS/ER stress mediators. These factors act as molecular switches that dynamically integrate cellular stress signals to determine cell fate between survival and death. A detailed understanding of these crosstalk factors and their context-dependent functions is critical for elucidating the pathogenesis of human tumors and developing novel therapeutic strategies that target the balance between autophagy and apoptosis.
Apoptosis mediated by death receptor
Apoptosis mediated by endoplasmic reticulum
Apoptosis mediated by mitochondria
Aesthetic Appreciation of Apoptosis Signal Pathway
References
[1] Cosford NDP. Cell Survival and Cell Death at the Intersection of Autophagy and Apoptosis: Implications for Current and Future Cancer Therapeutics [J]. ACS Chem Biol. 2021;16(12):2287-2297.
[2] Su, M., Mei, Y., & Sinha, S. (2013). Role of the Crosstalk between Autophagy and Apoptosis in Cancer [J]. Journal of Oncology, 2013, 102735.
[3] Yan, Y., Yu, W., et al. (2024). Autophagy regulates apoptosis of colorectal cancer cells based on signaling pathways [J]. Discover Oncology, 15, 367.
[4] Chakraborty S, Nandi P, et al. Molecular mechanisms in regulation of autophagy and apoptosis in view of epigenetic regulation of genes and involvement of liquid-liquid phase separation [J]. Cancer Lett. 2024 Apr 10;587:216779.
[5] Palabiyik, A. A. (2025). The role of Bcl‑2 in controlling the transition between autophagy and apoptosis (Review) [J]. Molecular Medicine Reports, 32(1), 172.
[6] Levine, B., Sinha, S., & Kroemer, G. (2008). Bcl-2 family members: Dual regulators of apoptosis and autophagy [J]. Autophagy, 4(5), 600.
[7] Young, M. M., Kester, M., & Wang, H. G. (2012). Sphingolipids: Regulators of crosstalk between apoptosis and autophagy [J]. Journal of Lipid Research, 54(1), 5.
[8] Bata, N., & P Cosford, N. D. (2021). Cell Survival and Cell Death at the Intersection of Autophagy and Apoptosis: Implications for Current and Future Cancer Therapeutics [J]. ACS Pharmacology & Translational Science, 4(6), 1728.
[9] Amaravadi, R. K., Kimmelman, A. C., & Debnath, J. (2019). Targeting Autophagy in Cancer: Recent Advances and Future Directions [J]. Cancer Discovery, 9(9), 1167.
[10] Wu, H., Che, X., et al. (2014). Caspases: A Molecular Switch Node in the Crosstalk between Autophagy and Apoptosis [J]. International Journal of Biological Sciences, 10(9), 1072.
[11] Mukhopadhyay S, Panda PK, Sinha N, Das DN, Bhutia SK. Autophagy and apoptosis: where do they meet [J]? Apoptosis. 2014 Apr;19(4):555-66.
[12] Yonekawa, T., & Thorburn, A. (2013). Autophagy and Cell Death [J]. Essays in Biochemistry, 55, 105.
[13] Galluzzi, L., Bravo-San Pedro, J. M., Kepp, O., & Kroemer, G. (2016). Regulated cell death and adaptive stress responses [J]. Cellular and Molecular Life Sciences: CMLS, 73(11-12), 2405.
[14] Jung, C. H., Ro, S. H., Cao, J., Otto, N. M., & Kim, D. H. (2010). MTOR regulation of autophagy [J]. FEBS Letters, 584(7), 1287.
[15] Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis [J]. Nat Rev Mol Cell Biol. 2007 Sep;8(9):741-52.
[16] Sukumaran, P., Da Conceicao, V. N., et al. (2021). Calcium Signaling Regulates Autophagy and Apoptosis. Cells, 10(8), 2125.
[17] Yu, Y. S., Kim, I. S., & Baek, S. H. (2025). Decoding the dual role of autophagy in cancer through transcriptional and epigenetic regulation [J]. Febs Letters, 599(16), 2237.
[18] Jain, V., Singh, M. P., & Amaravadi, R. K. (2023). Recent advances in targeting autophagy in cancer [J]. Trends in Pharmacological Sciences, 44(5), 290.
[19] Brun, S., Bestion, E., et al. (2021). GNS561, a clinical-stage PPT1 inhibitor, is efficient against hepatocellular carcinoma via modulation of lysosomal functions [J]. Autophagy, 18(3), 678.
[20] Walweel, N., & Aydin, O. (2024). Enhancing Therapeutic Efficacy in Cancer Treatment: Integrating Nanomedicine with Autophagy Inhibition Strategies [J]. ACS Omega, 9(26), 27832.
[21] Ibrahim Abdelrahman Hassan, A. M., Zhao, Y., Chen, X., & He, C. (2024). Blockage of Autophagy for Cancer Therapy: A Comprehensive Review [J]. International Journal of Molecular Sciences, 25(13), 7459.
[22] Townsend, P. A., Kozhevnikova, M. V., et al. (2021). BH3-mimetics: Recent developments in cancer therapy [J]. Journal of Experimental & Clinical Cancer Research: CR, 40, 355.
[23] Saxena, R., Welsh, C. M., & He, Y. W. (2024). Targeting regulated cell death pathways in cancers for effective treatment: A comprehensive review [J]. Frontiers in Cell and Developmental Biology, 12, 1462339.
[24] Varkaris, A., Wang, K., et al. (2025). BH3 mimetics targeting BCL-XL have efficacy in solid tumors with RB1 loss and replication stress [J]. Nature Communications, 16(1), 4931.
[25] Kuma, A., Hatano, M., et al. (2004). The role of autophagy during the early neonatal starvation period [J]. Nature, 432(7020), 1032-1036.
[26] Ogata, M., Hino, S. I., et al. (2006). Autophagy Is Activated for Cell Survival after Endoplasmic Reticulum Stress [J]. Molecular and Cellular Biology, 26(24), 9220.
[27] Wirawan, E., Walle, L. V., et al. (2010). Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria [J]. Cell Death & Disease, 1(1), e18.


Comments
Leave a Comment