Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
In 1906, Dr. Alzheimer observed distinctive plaques and neurofibrillary tangles in the brain tissues of a woman who suffered from memory loss, language problems, and unpredictable behaviors during her illness until her death. This was the first description of Alzheimer's disease. In 1910, Krepelin named the neurological condition “Alzheimer's disease” in the eighth edition of “Psychiatry”. Alzheimer's has since become widely known around the world.
1. What Is Alzheimer's Disease?
2. What Are the Symptoms of Alzheimer's Disease?
4. Data about Alzheimer's Disease
Alzheimer's disease (AD) is the prevailing type of dementia, constituting 60% to 70% of dementia cases globally [1]. It is a progressive neurodegenerative disorder that impairs the ability to memory, think, learn, and organize, ultimately severely affecting social, occupational and life functions. It generally occurs in old age and early senility.
Neuropathologically, AD is characterized by significant cortical atrophy and enlargement of ventricular. Pathological hallmarks are the senile plaques formed by the abnormal buildup of amyloid-β (Aβ) protein, and neurofibrillary tangles of hyperphosphorylated tau (t) protein.
Alzheimer's disease started insidiously and the course of the disease was chronic progressive. The symptoms of Alzheimer's disease vary from person to person. However, it shares some common symptoms in the early stage, including progressive memory impairment, cognitive dysfunction, language barrier, movement disorder, and personality and behavior changes, as well as mental disorders [2].
These symptoms gradually deteriorate over time. In the advanced stages of the disease, the patients develop serious complications, such as dehydration, malnutrition, or infection, which can eventually lead to death [3]. .
The exact cause of Alzheimer's disease is not fully understood, and it is likely influenced by a combination of genetic, environmental, and lifestyle factors.
Most cases of Alzheimer's disease are not directly inherited, but certain genetic factors can increase the risk. Mutations in specific genes, such as APP (amyloid precursor protein)[4], PSEN1 (presenilin 1), and PSEN2 (presenilin 2), are associated with familial forms of Alzheimer's disease and occur at early-onset AD (EOAD). Individuals with these mutations have a higher likelihood of developing Alzheimer's disease at an earlier age.
The APOE gene is the most well-established genetic risk factor for late-onset AD (LOAD). The majority of LOAD cases occur sporadically, with no reported family history of the condition. A substantial number of genes identified by the genome-wide association study (GWAS) could be associated with the Aβ cascade or tau pathology, and tend to cluster within three distinct pathways.
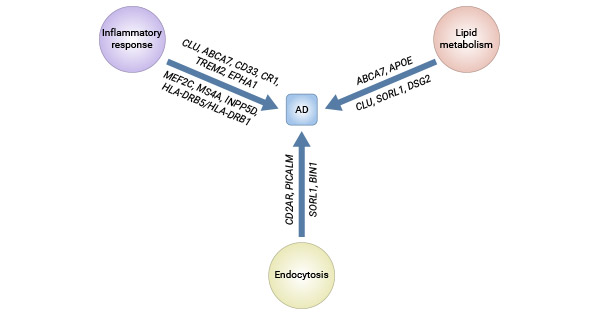
Figure 1. Major pathways involved in AD and affected genes
This picture is cited from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4876682/
| Alzheimer's disease associated genes | Function | |
|---|---|---|
| Early-onset AD (EOAD) Genes | APP | Neuronal development, Synaptic formation and repair, β-Amyloid production |
| PSEN1 | γ-Secretase activity, Intracellular signaling, APP processing, β-Amyloid production | |
| PSEN2 | γ-Secretase activity, APP processing, β-Amyloid production, Synaptic plasticity | |
| amyloid-β(Aβ) | Plaque formation | |
| tau | Neurofibrillary tangle formation | |
| BACE1 (beta-secretase) | Cleave APP to release the sAPPβ and generate C99 in the first step of the amyloidogenic pathway | |
| gamma-secretase | Cleave C99 to release beta-amyloid peptides in the second step of the amyloidogenic pathway | |
| Late-onset AD (LOAD) Genes | Apolipoprotein E (ApoE) | Individuals with one copy of the APOEε4 allele have an increased risk, and those with two copies have an even higher risk |
| CLU | Synapse turnover Complement regulation Chaperone protein |
|
| ABCA7 | Phagocytosis Lipid homeostasis |
|
| SORL1 | Endocytosis Receptor for APOE APP trafficking |
|
| CR1 | Amyloid β clearance Complement activation |
|
| CD33 | Clathrin-mediated endocytosis Cell signaling |
|
| MS4A | Signal transduction Immune function |
|
| TREM2 | Inflammatory response | |
| BIN1 | Synaptic vesicle endocytosis APP trafficking Cytoskeletal dynamics |
|
| CD2AP | Receptor-mediated endocytosis Cytokinesis Cytoskeletal dynamics |
|
| PICALM | Clathrin-mediated endocytosis | |
| EPHA1 | Synaptic development Immune function Neural development |
|
| HLA-DRB5/HLA-DRB1 | Immune function Histocompatibility |
|
| INPP5D | Cytokine signaling Immune function |
|
| MEF2C | Myogenesis Synapse formation |
|
| CASS4 | Cell migration Cell adhesion |
|
| PTK2B | Calcium homeostasis MAP kinase signaling |
|
| NME8 | Ciliary function Neuronal cell proliferation |
|
| ZCWPW1 | Epigenetic regulation Neural development |
|
| CELF1 | mRNA editing Pre-mRNA splicing |
|
| FERMT2 | Cell–cell adhesion Angiogenesis |
|
| SLC24A4/RIN3 | Cell signaling Neural development |
|
| DSG2 | Cell–cell adhesion | |
| PLD3 | Signal transduction Epigenetic modification |
|
| UNC5C | Neural development | |
| AKAP9 | Signal transduction | |
| ADAM10 | Hippocampal neurogenesis Cell adhesion |
|
The Table information is cited from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4876682/
Biological Characteristics of Alzheimer's Disease:
The characteristic feature of Alzheimer's is the accumulation of extracellular amyloid β (Aβ) plaques and the presence of intracellular neurofibrillary tangles (NFTs) containing hyper-phosphorylated tau [5]. The aggregation of beta-amyloid proteins can disrupt communication between neurons and contribute to neuronal damage. Tau protein is essential for maintaining the structure of neurons, and abnormal aggregation leads to the formation of tangles, disrupting neural function.
These changes are toxic to neurons, resulting in neural dysfunction and inter-connection loss and death. The resulting lesions initially occur in areas of the brain responsible for memory including the entorhinal cortex and hippocampus causing language, behavior, and logic problems, and then spread to almost the whole brain by the last stage of Alzheimer's, leading to significant brain atrophy.
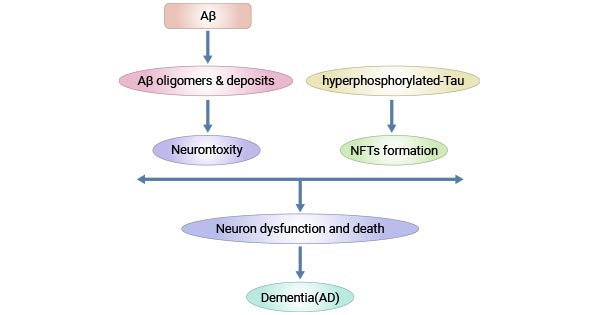
Figure 2. Pathogenesis of Alzheimer's disease
This picture id cited from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4588032/
At the 2017 Alzheimer's Association International Conference (AAIC), a report from the Lancet Commissions [6] caused extensive discussion. The report entitled Dementia Prevention, Intervention, and Care presents nine risk factors for dementia. Based on the population attribution score (PAF), the following major risk factors were identified:
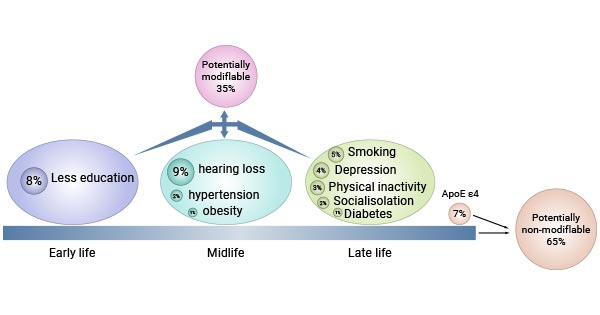
Figure 3. Effects of modifiable and non-modifiable risk factors on Alzheimer's disease
As of 2020, extensive research has revealed additional risk factors for Alzheimer's disease and dementia, including excessive alcohol intake, brain damage, and air pollution [7].
Alzheimer's disease was officially recognized as the sixth leading cause of death in the United States in 2019. In 2020 and 2021, when COVID-19 became one of the top ten causes of death, it fell to seventh place. About 6.7 million Americans aged 65 and older live with Alzheimer's disease in 2023. This number is expected to rise to 13 million by 2050. It is estimated that 60% to 70% of the approximately 55 million people worldwide with dementia have Alzheimer's disease.
Early onset (before 65 years old) of Alzheimer's disease accounted for only about 5% of all Alzheimer's patients. Alzheimer's patients mostly are late-onset (after 65 years of age), and more common in people over 70 years old. Demographic epidemiological statistics indicate that there are very few people with Alzheimer's disease in people under the age of 40 [8]. As early as 2011, the global incidence of dementia reached about 24 million, and this number is still growing at a high speed. It is expected to double every 20 years by 2040 [9,10].
Epidemiological data indicates that in the United States, one out of every three individuals aged over 85 is projected to develop Alzheimer's disease. Furthermore, by the year 2050, the population of Americans aged over 85 years is expected to triple. According to the Global Burden of Disease Study, approximately 57.4 million people globally were living with dementia in 2019. This number is expected to reach 152.8 million people by 2050, which is three times that of the present.
Alzheimer's disease is a complex neurodegenerative disorder with multiple underlying molecular mechanisms. Several signal pathways are implicated in the pathogenesis of Alzheimer's disease.
Recent studies have found that the c-jun N-terminal kinase signal transduction pathway was activated in both the brain of the animal model of transgenic Alzheimer's disease and the brain of patients with Alzheimer's disease, and believed that this pathway was related to the deposition of Aβ [11,12]. JNK signal transduction pathway is also closely associated with another pathological change in Alzheimer's disease - abnormal phosphorylation of Tau protein and formation of double-stranded filaments [13]. All three pathways of MAPK - ERK, JNK, and p38, are involved in the induction of tau hyperphosphorylation and are related to factors such as Aβ, oxidative stress, inflammatory factors, and protein phosphatase.
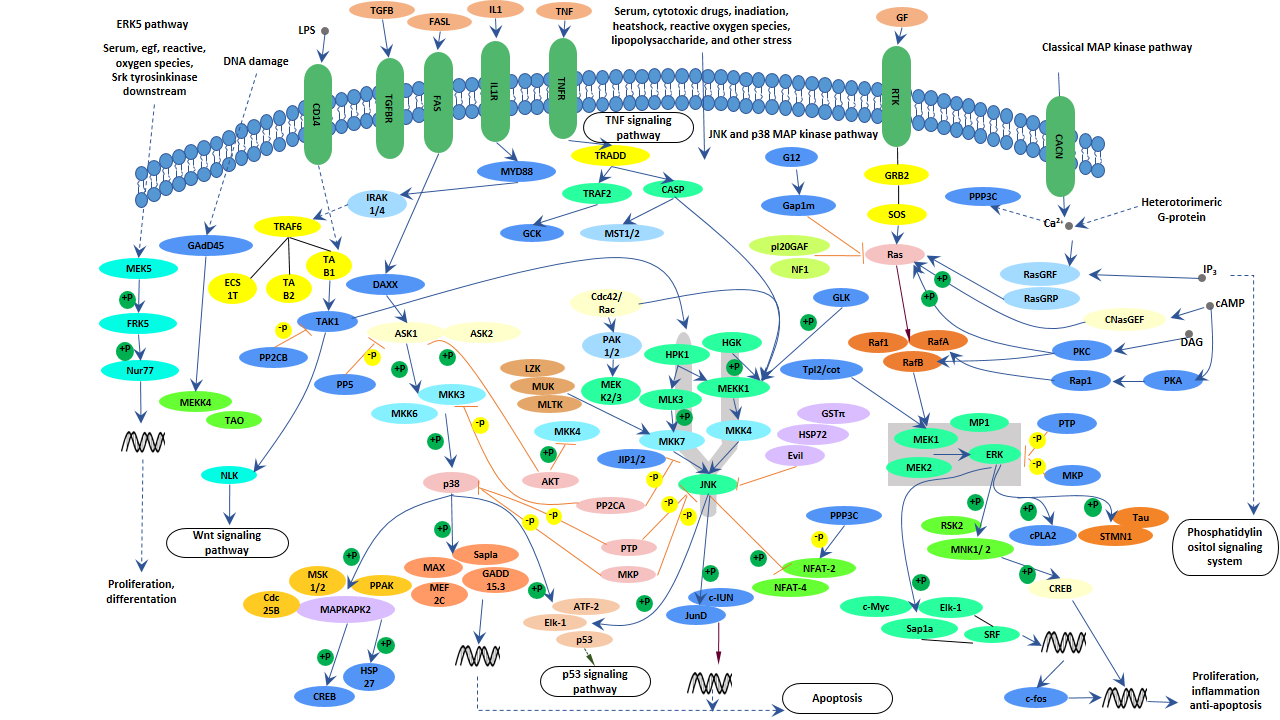
Figure 4. MAPK Signaling Pathway
The mTOR pathway is an important regulatory pathway for neuronal development. Its functions include the following aspects:
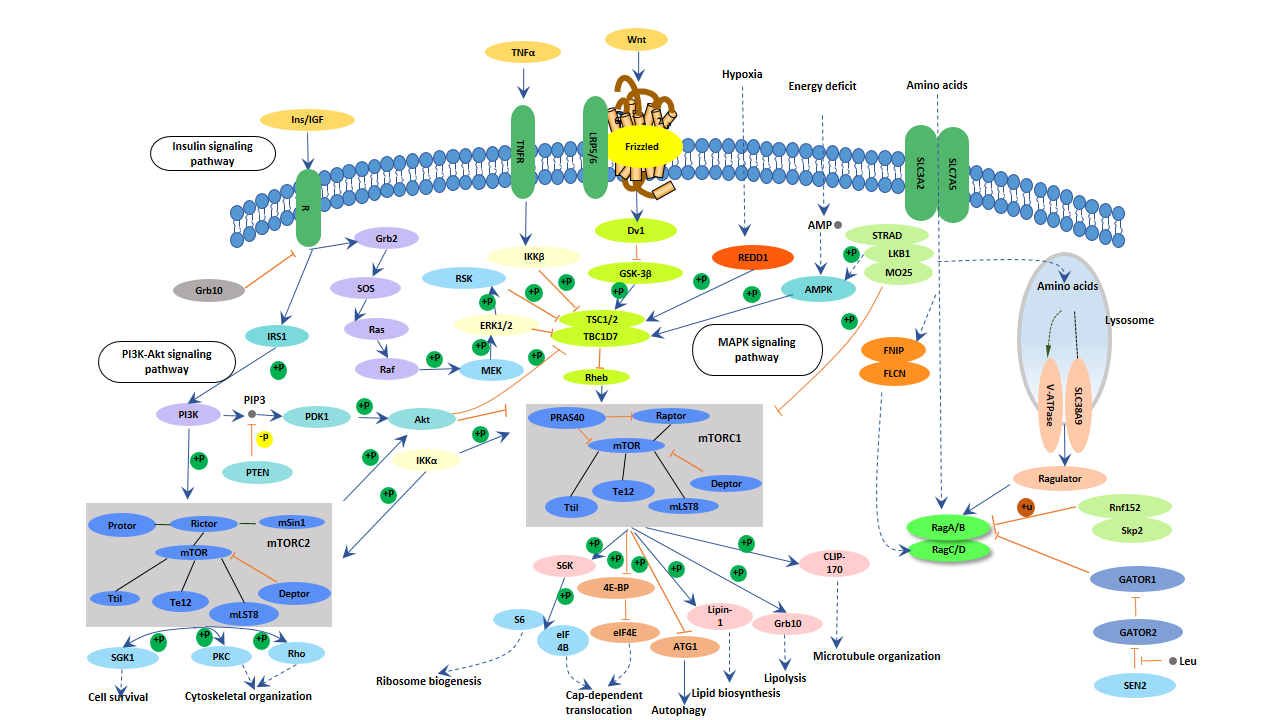
Figure 5. mTOR signaling pathway
One characteristic of Alzheimer's disease is the large number of neuronal loss in certain areas of the brain, which may be related to abnormalities in the mTOR pathway. There is ample evidence that mTOR can influence the formation of learning and memory through different molecular mechanisms. Activation of the mTOR pathway directly up-regulates proteins involved in synaptic plasticity regulation, affecting learning and memory. The abnormal mTOR pathway in Alzheimer's disease (AD) patients also suggests that the regulation of mTOR signaling is an important factor for neurological disease [14].
Insulin receptors are widely distributed in the brain, including the hippocampus, olfactory bulb, and hypothalamus. There are three main insulin signaling pathways in the brain: insulin PI3K/AKT-GSK3 signaling pathway, insulin PI3K/AKT-BAD signaling pathway, and insulin PI3K/AKT-mTOR signaling pathway. Insulin in neuronal cells accelerates the production of neurons through MAPK and PI3K/AKT. When the insulin signaling pathway fails, the PI3K/AKT and MAPK signaling pathway are inhibited, resulting in apoptosis. Insulin can also regulate N-methyl-D-aspartate (NMDA) expression and long-term potent action (LTP) excitability, which in turn affects memory and learning ability. A large number of studies have shown that pathological changes in AD are associated with neuronal insulin receptor (IR) signal transduction pathway disorders [15].
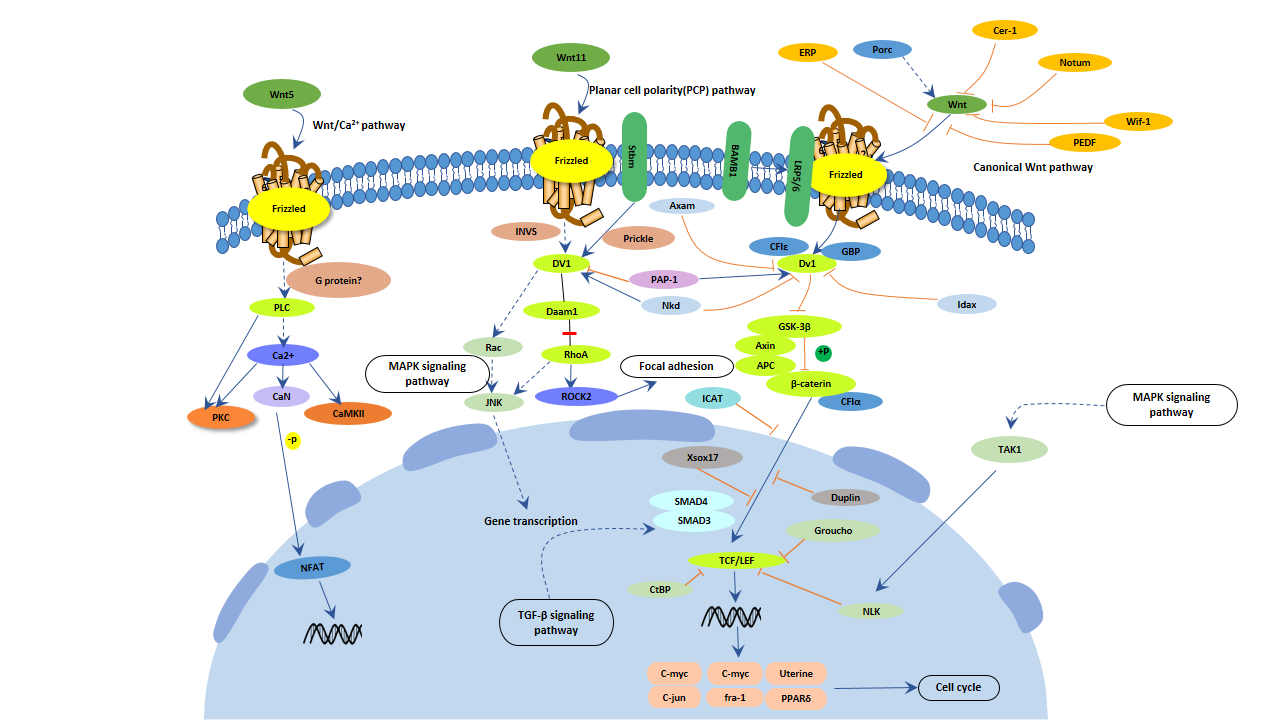
Figure 6. Wnt signaling pathway
The wnt signaling pathway is involved in most of the processes of functional intact neurons, and numerous studies have shown that the wnt signaling pathway plays an important role in the development of Alzheimer's disease [16].
Although there is currently no cure for Alzheimer's disease, numerous promising research areas are being investigated in the hopes of finding new treatments and prevention strategies. Immunotherapy and biomarker development stand out among these current hot research fields of Alzheimer's disease.
Immunotherapies for AD primarily focus on targeting beta-amyloid. Toxic extracellular deposition of misfolded beta-amyloid protein aggregate is a hallmark of AD. The Aβ-based immunotherapy primarily aims to reduce Aβ aggregate formation, spread, and deposition in the human brain. The two main anti-Aβ therapy concepts are active and passive immunization. Passive vaccination directly introduces exogenous monoclonal antibodies (mAbs), while active vaccination directs the immune system to produce an immunological response using an anti-Aβ agent. Anti-Aβ agents have so far focused on lowering Aβ generation, suppressing Aβ aggregation, and accelerating Aβ clearance.
Conventional Alzheimer's disease (AD) biomarkers play a crucial role in aiding clinicians in the diagnosis of Alzheimer's disease, assessing disease progression, and monitoring the effects of potential therapeutic interventions.
Here are some of the commonly used biomarkers:
| Biomarkers | Effects | |
|---|---|---|
| Beta-Amyloid (Aβ) Peptides | CSF Aβ42 | Reduced levels of Aβ42 in the cerebrospinal fluid (CSF) are associated with amyloid plaque deposition in the brain |
| Amyloid PET Imaging | Positron emission tomography (PET) scans using radiolabeled tracers can visualize and quantify amyloid plaques in the brain | |
| Tau Proteins | CSF Tau | Elevated levels of total tau (t-tau) and phosphorylated tau (p-tau) in the CSF are indicative of neurofibrillary tangle formation and neurodegeneration |
| Tau PET Imaging | PET imaging with tau tracers allows for the visualization of tau pathology in the brain | |
| Neurodegeneration Biomarkers | CSF Neurofilament Light (NfL) | Elevated NfL levels in the CSF indicate neurodegeneration and axonal damage |
| Structural Brain Imaging | Magnetic resonance imaging (MRI) can reveal structural changes in the brain associated with neurodegeneration | |
| Imaging Biomarkers | Functional Brain Imaging (FDG-PET) | Fluorodeoxyglucose PET measures glucose metabolism in the brain, providing information about neuronal activity |
| MRI-Based Volumetric Measures | Changes in brain volume, particularly in regions like the hippocampus, are associated with AD progression | |
| Cognitive Biomarkers | Cognitive Testing | Assessments of memory, executive function, and other cognitive domains help evaluate the severity of cognitive impairment |
| Neurofilament Light Chain (NfL) in Blood | Elevated levels of NfL in blood may reflect neurodegeneration | |
However, the invasiveness of CSF-based measurements and the high costs of PET imaging render these tests less appealing and time-consuming. Consequently, it is critically important to explore non-invasive and precise biomarkers that can indicate the risk of developing AD and classify its various stages.
Leveraging recent technological advancements, the scientific community has explored/ put forward various biomarkers in blood, plasma, serum, urine, and saliva to detect AD early before LOAD [17, 18]. The omics technologies are capable of profiling multiple molecules simultaneously, which can avoid the disadvantage of heterogeneity among patients. The combination of molecular patterns and computational power, including artificial intelligence (AI) and machine-learning (ML) tools, holds promise for addressing these challenges [19].
Researchers are investigating AI-based methods for diagnosing AD using diverse modalities such as biofluid biomarkers, retinal and iris readings, electroencephalogram (EEG) brain wave measurements [20], and online language skills and memory tests [21,22]. These advancements could potentially facilitate mass screening for AD, provided they demonstrate high accuracy in real clinical settings.
miRNAs have also been detected in plasma, serum, and CSF as markers for various diseases, including cardiovascular diseases, cancer, and neurodegenerative disorders [23]. Studies indicate that nearly 50% of known miRNAs are expressed in the nervous system, playing a significant role in regulating normal brain physiology, aging, and mental health. In Alzheimer's disease, miRNAs target crucial disease-related genes, demonstrating either neurodegenerative or neuroprotective effects [24].
These solutions represent significant advancements, but there is ongoing debate and exploration regarding their effectiveness, long-term outcomes, and potential side effects.
References
[1] Oh, E. S., and Rabins, P. V. (2019). Dementia [J]. Ann. Intern. Med. 171, ITC33–ITC48.
[2] Lyketsos, C. G., Carrillo, et al. (2011). Neuropsychiatric symptoms in Alzheimer's disease [J]. Alzheimer's Dement. 7, 532–539.
[3] Bird, T. D. Alzheimer disease overview. GeneReviews®[Internet] (2018).
[4] Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing and function [J]. J Biol Chem. 2008;283(44):29615–29619.
[5] Querfurth H W, Laferla F M. Alzheimer's disease [J]. N Engl J Med, 2010, 362(4):329-344.
[6] Livingston G, Sommerlad A, Orgeta V,et al. Dementia prevention, intervention, and care [J]. Lancet, 2017, 390(10113).
[7] Livingston, G., Huntley, J., et al. (2020). Dementia prevention, intervention, and care: 2020 report of the Lancet Commission [J]. Lancet 396, 413–446.
[8] Harvey R, Skelton-Robinson M, Rossor M. The prevalence and causes of dementia in people under the age of 65 years [J]. Journal of Neurology Neurosurgery & Psychiatry, 2003, 74(9):1206.
[9] Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease [J]. Nature Reviews Neurology, 2011, 7(3):137-152.
[10] Chan K Y, Wang W, Wu J J, et al. Epidemiology of Alzheimer's disease and other forms of dementia in China, 1990–2010: a systematic review and analysis [J]. The Lancet,2013, 381(9882): 2016-2023.
[11] Savage M J, Lin Y G, Ciallella J R, et al. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer's disease model is associated with amyloid deposition [J]. Journal of Neuroscience the Official Journal of the Society for Neuroscience, 2002, 22(9):3376.
[12] Shoji M, Iwakami N, Takeuchi S, et al. JNK activation is associated with intracellular beta-amyloid accumulation [J]. Molecular Brain Research, 2000, 85(1-2):221-233.
[13] CristianaAtzori, BernardinoGhetti, RobertoPiva, et al. Activation of the JNK/p38 Pathway Occurs in Diseases Characterized by Tau Protein Pathology and Is Related to Tau Phosphorylation But Not to Apoptosis [J]. J Neuropathol Exp Neurol, 2001, 60(12):1190-1197.
[14] Lafay-Chebassier C, Paccalin M, Page G, et al. mTOR/p70S6k signalling alteration by A-beta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer's disease [J]. Journal of Neurochemistry, 2005, 94(1):215-225.
[15] Grimm MO1, Hundsdörfer B, Grösgen S, et al. PS dependent APP cleavage regulates glucosylceramide synthase and is affected in Alzheimer's disease [J] . Cell Physiol Biochem, 2014, 34: 92-110.
[16] Boonen R A, Van T P, Zivkovic D. Wnt signaling in Alzheimer's disease: up or down, that is the question [J]. Ageing Research Reviews, 2009, 8(2):71-82.
[17] Cheng S., Banerjee S., et al. Novel blood test for early biomarkers of preeclampsia and Alzheimer's disease [J]. Sci. Rep. 2021;11:15934.
[18] Goldoni R., Dolci C., et all. Salivary biomarkers of neurodegenerative and demyelinating diseases and biosensors for their detection. Ageing Res. Rev. 2022;76:101587.
[19] Borhani N., Ghaisari J., Abedi M., Kamali M., Gheisari Y. A deep learning approach to predict inter-omics interactions in multi-layer networks [J]. BMC Bioinform. 2022;23:53.
[20] Meghdadi A.H., Stevanović Karić M., et al. Resting state EEG biomarkers of cognitive decline associated with Alzheimer's disease and mild cognitive impairment [J]. PLoS ONE. 2021;16:e0244180.
[21] Eyigoz E., Mathur S., et al. Linguistic markers predict onset of Alzheimer's disease [J]. EClinicalMedicine. 2020;28:100583.
[22] Gunes S, Aizawa Y, et al. Biomarkers for Alzheimer's Disease in the Current State: A Narrative Review [J]. Int J Mol Sci. 2022 Apr 29;23(9):4962.
[23] Silvestro S, Bramanti P, Mazzon E. Role of miRNAs in Alzheimer's disease and possible fields of application [J]. Int J Mol Sci. 2019;20(16):3979.
[24] Reddy PH, Tonk S, et al. A critical evaluation of neuroprotective and neurodegenerative MicroRNAs in Alzheimer's disease [J]. Biochem Biophys Res Commun. 2017;483(4):1156–65.


Comments
Leave a Comment