Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
DNA is the repository of genetic information that is necessary for an organism to survive and reproduce, so maintaining the integrity of DNA molecules is crucial to cells. However, DNA is inevitably damaged during its continuous copy and transmission to the daughter cells. So how do cells deal with damaged DNA? What is exactly DNA damage? What causes DNA damage? What diseases are DNA damage associated with?
In this review, DNA damage and its causes as well as its differences from mutation are outlined. Finally, types of DNA damage, DNA damage response pathways, and associated diseases are comprehensively described.
Damage to DNA refers to any alterations that introduce deviations from its original double-helical structure. DNA lesions occur in all cellular organisms.
There are many sources of DNA damage, which can be mainly divided into two categories: endogenous and exogenous factors. Cell-intrinsic DNA lesions include replication errors and spontaneous genotoxic insults arising from endogenous chemical reactions. External sources can be subdivided into physical and chemical factors.
Every cell during its lifespan must constantly fight against endogenous and exogenous DNA insults [1].
It has been estimated that each cell experiences almost 100,000 spontaneous DNA damage per day [2].
DNA replication based on base pairing with DNA as a template is a strict and precise event, but it is not completely error-free. The frequency of base-pairing errors is about 10-1-10-2. Under the action of DNA replicase, the frequency of base-pairing errors is reduced to about 10-5-10-6. If there is an incorrect nucleotide involved in the replication process, DNA polymerase will also suspend its catalysis. Although the probability of base mismatches during replication is very low, there are still some mismatched bases that escape proofreading and even MMR mechanisms. When the template is damaged, the trans-damage synthesis (TLS) polymerase has low fidelity and is an important source of spontaneous mutations. In addition, there are errors caused by topoisomerase, uracil incorporation, and so on.
Although most DNA replicates with fairly high fidelity, errors can sometimes occur. Insertion and deletion of nucleotide bases are possible happenings. Besides, spontaneous errors during the replication of DNA might lead to the incorporation of wrong nucleotides in the nascent synthesized DNA molecule, resulting in mismatched base pairs [3].
Hydrolytic damage to DNA involves deamination and loss of individual bases. Biochemical reactions of various metabolites and excessive reactive oxygen species (ROS) may contribute to hydrolytic insults.
● Base deamination
Deamination of nitrogenous bases is containing amino groups will spontaneously deamination, resulting in C-U, A-I (hypoxanthine), and G-X (xanthine). Interestingly, the frequency of base deamination in single-stranded strands is much higher than that in double-stranded strands, so the transient single-stranded state during replication, transcription, and recombination will exacerbate this damage, and it will occur before repair Mutation effect. Among all bases, 5-methylcytosine has the highest frequency of deamination and the generated G: T base pair is corrected by the slower mismatch repair (MMR). Scattered CpG sequences are easily affected by cytosine 5-methyltransferase. About 70%-80% of vertebrate CpG cytosines are methylated. The GC→AT base conversion caused by its deamination accounts for one-third of the point mutations in human genetic diseases.
● Base Loss
DNA base loss, also known as AP (apurinic/apyrimidinic) sites, can be particularly mutagenic and if left unrepaired they can inhibit transcription. Bases can also be lost. E. coli loses about one purine per generation, and mammalian cells lose about 10,000 per day. Relatively speaking, the N-glycosidic bond of pyrimidine is relatively stable, and the probability of loss is only 1/20 of that of purine. Abasic sites are unstable and are prone to β-elimination reactions to cause single-strand breaks (SSB).
● Base tautomerism
The isomers of the four bases in DNA can all change spontaneously (for example, the interconversion between enol and keto bases). This change will change the hydrogen bond between base pairings. Adenine can be matched with cytosine, thymine can be matched with guanine, etc. If these pairings occur during DNA replication, it will cause erroneous damage in the offspring's DNA sequence that is different from that of the parent's DNA. The bases have spontaneous tautomerism of ketone and enol structures, which can cause base mismatches. For example, when adenine forms an A=NH structure, it can form A=C pair, and when guanine forms a G-OH structure, it can form a GT triple bond pairing.
Reactive oxygen species (ROS) can also cause DNA damage. Active oxygen free radicals can attack double bonds on bases, causing ring-opening reactions, etc., and can also damage the ribose phosphate backbone and cause single-strand breaks. This is similar to ionizing radiation. It is estimated that reactive oxygen species can cause 2,300 single-strand breaks per hour in mammalian cells. ROS-producing DNA damage is the most frequently occurring damage.
Environmental DNA damage can be generated by physical or chemical sources.
Common physical factors mainly include ionizing radiation (IR) and solar ultraviolet (UV) light.
IR in daily life commonly comes from cosmic radiation, X-rays, exposure to radioactive substances, and medical treatments using radiotherapy. It can induce base modifications, interstrand crosslinks, and DNA strand breaks, especially double-strand breaks (DSBs). In addition, IR also induces the formation of ROS, which further damages DNA.
UV light from the sun can react with DNA and mainly causes two adjacent pyrimidines to form dimers, hindering DNA replication and transcription.
IR includes X-rays, gamma rays, alpha and beta particles, and neutrons. IR in daily life commonly comes from cosmic radiation and medical treatments using X-rays or radiotherapy. It directly affects DNA structure by producing DNA breaks, especially double-strand breaks (DSBs). IR also causes the formation of reactive oxygen species (ROS), which may generate apurinic/apyrimidinic (abasic) sites, single strand breaks (SSBs), sugar moiety modifications, and deaminated adducted bases [4] [5].
The chemical events that lead to DNA damage include hydrolysis, exposure to reactive oxygen substances (ROS) and other reactive metabolites.
As relatively common metabolic byproducts, ROS can cause single- and double strand breaks, aducts, and crosslinks.
There are many chemical factors that can cause DNA damage. The alkylating agent has an active alkyl group, which can be transferred to the base or phosphoric acid, such as dimethyl sulfate, methyl methanesulfonate (MMS), mustard gas, etc. The O6 and N7 of guanine are most easily alkylated, leading to mismatch (GT) or shedding. Phosphoric acid triesters are unstable and easy to break. The bifunctional alkylating agent can cause crosslinking and is called a crosslinking agent. Certain alkylating agents such as cyclophosphamide (cyclophosphamide) can be used in chemotherapy. Base or nucleoside analogs, such as 5-fluorouracil (FU), 5-bromouracil (BrdU), 6-mercaptopurine, etc., can compete to inhibit nucleotide synthesis or incorporate nucleic acid to cause mismatches. Nitrite can cause base deamination, and nitrosamines are oxidized to generate alkylating agents and free radicals. There is also a class of chemical substances called metabolic activating compounds, which are catalyzed by the liver's mixed functional oxidase (cytochrome P450) to form active alkylating agents or epoxides, etc., which interact with nucleic acids and cause mutations. Such as aromatic amines, polycyclic aromatic hydrocarbons (PAH), etc. Benzopyrene is one of the most carcinogenic polycyclic aromatic hydrocarbons. Tobacco smoke contains a lot of aromatic amines and polycyclic aromatic hydrocarbons. Aflatoxin is also a metabolically activated carcinogen, and aflatoxin B1 has the strongest effect. After passively diffused into the cell, aflatoxin B1 is metabolized by the P-450 complex to the active form of aflatoxin B1-8,9-epoxide, which is then added to guanine N7 to weaken the glycosidic bond and lead to depurination.
Although DNA damage and DNA mutation are both types of DNA errors, they are significantly different from each other. DNA damages are physical abnormalities in DNA such as single- and double-strand breaks, while a mutation is a change in the base sequence of the DNA. DNA damages can be recognized by enzymes and thus correctly repaired if redundant information is available for copying. While most DNA damages can undergo DNA repair, the unrepaired DNA damages can accumulate in replicating cells, giving rise to mutations. In contrast to DNA damage, DNA mutations can not be recognized and repaired by enzymes once the base change is present in both DNA strands, which can cause alterations in protein function and regulation.
The main difference between DNA damage and DNA mutation is that DNA damage is a structural change in DNA whereas mutation is a change in the nucleotide sequence of DNA. Furthermore, DNA damage prevents DNA replication while mutation alters the genetic information encoded by DNA. DNA damage and mutation are groups of errors that occur in DNA. Moreover, environmental factors, as well as the compound released by metabolism, can cause DNA damage while mutations mainly arise due to the errors in DNA replication and recombination.
All living cells inevitably undergo different types and degrees of DNA lesions. To survive these unfavorable conditions and faithfully pass on intact genetic information to the next generation, cells have evolved a highly ordered and coordinated mechanism known as DNA damage response (DDR) to attenuate these genotoxic stress.
DDR is a key factor involved in sustaining genome integrity and stability. DDR sensor proteins recognize damaged DNA and initiate a cascade of signal transducers (such as ATM, ATR, or the Rad17-RFC complex), which lead to the activation of DDR mediators (such as serine/threonine kinase Chk1, Chk2, Cdc25 phosphatases). Activated DDR mediators enable the activation of specific protein machinery, triggering an appropriate cellular response, which could be DNA repair, cell cycle checkpoint activation, induction of apoptosis or senescence, or tolerance of lesions [6-9]. This varying response depends on the cell type, the extent of damage, and other factors.
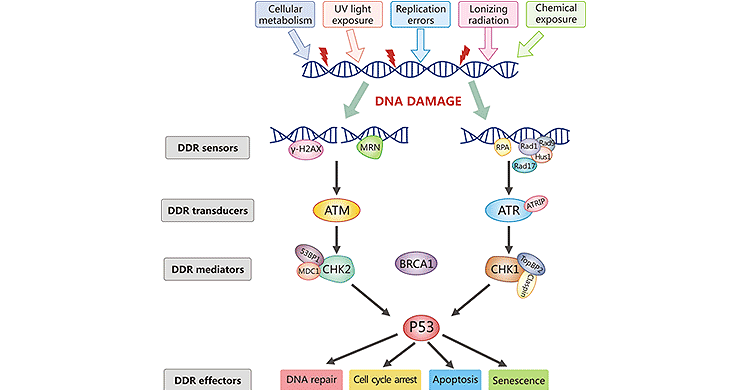
Figure: DNA Damage Response Signaling Pathway
*The picture is cited from https://www.sciencedirect.com/science/article/abs/pii/S156878641830096X
The activation of the DNA damage repair system is a major end-point of DNA damage response. Eukaryotic cells exhibit several DNA repair mechanisms to deal with potential DNA lesions [10]. These repair pathways have varying fidelity and mutagenic consequences [10]. These mechanisms sense and recognize all types of DNA damage, suspend genome replication, signal for repair, and correct or tolerate DNA lesions [11]. There is very little redundancy between the different repair mechanisms.
DNA repair mechanisms include direct repair, base excision repair, nucleotide excision repair, double-strand break repair, and cross-link repair.
Excision repair mechanisms targeted to the removal of bulky DNA adducts and UV-induced photoproducts, base-pair alterations and purine loss, and DNA mismatches, and single- and double-strand DNA breaks.
● Nucleotide Excision Repair (NER)
NER, the most versatile DNA repair pathway, can fix many types of physical and chemical factors-causing DNA insults, such as the UV-irradiation-mediated and cancer-causing chemical adducts to DNA. NER can excise large fragments of DNA damage. It can repair the forms of DNA damage, including 6-4 photoproducts, cyclobutane pyrimidine dimers, and bulky adducts.
● Base Excision Repair (BER)
BER is a mechanism that particularly helps remove small base modifications. It can repair those forms of oxidative, deamination, alkylation, and abasic single base damage that generally do not cause significant distortions to the DNA helix. In the BER pathway, DNA glycosylases detect and eliminate the impaired bases to form sites that are subsequently cleaved away by AP endonucleases, leaving behind a one-bas gap. The gap is further fixed by the DNA polymerase and DNA ligase.
The MMR pathway can correct mispaired bases, including base mismatch, insertion, and deletion. This repair system helps to identify DNA helix distortions, distinguish between the two strands, and get rid of replication mistakes. It increases the replication fidelity by more than 100 times through the correction of infrequent errors that have eluded replicative polymerases' proofreading [12-14].
DNA DSBs are the most cytotoxic DNA insults. DNA DSB repair pathways include homologous recombination (HR), DNA end-joining, and non-homologous end joining (NHEJ).
● Homologous Recombination (HR)
HR is an accurate repair pathway that requires a homologous DNA sequence as a repair template [15] and is restricted to S/G2 phases of the cell cycle [16]. HR is slow, complicated and highly accurate repair process involving numerous enzymes and proteins. This pathway can repair both one- and two-ended breaks and protein-blocked ends.
● Non-homologous end-joining (NHEJ)
NHEJ is an error-prone repair pathway predominantly used to rejoin DSBs in most mammalian cells. The occurring of NHEJ shows a preference during the G0, G1, and the early S phases [17-19]. It is the first line of defense against DSBs in mammalian cells before DNA replication. Compared to HR, NHEJ proceeds rapidly and is template- and cell cycle-independent. It is often mutagenic and can only repair two-ended breaks and can not repair protein-blocked ends. NHEJ involves ligation of break DNA ends and does not require sequence homology.
The alternative end-joining (a-EJ) pathway, which repairs DNA double-strand breaks (DSBs), is initiated by end resection that generates 3'single strand.
If damaged DNA is completely and timely fixed, there is almost little impact on cells. Unrepaired DNA may encode proteins necessary for the cell cycle, which leads to cell cycle arrest. The cell cycle is essential for cell growth, proliferation, and reproduction. It eventually influences the growth and survival of cells. If the damage is excessive, the cells no longer expend energy to repair the damage effectively and are likely to undergo apoptosis or senescence.
An accurate readout of DNA sequences is indispensable for the expression of functional messenger RNAs and ultimately proteins in cells. Besides, faithful DNA replication during cell division is essential for the daughter cells to inherit intact genetic materials from the parent cells. So various modifications of cellular DNA induced by intrinsic and internal factors can elicit profound biological consequences. Although most DNA damage can be repaired, the repair system is not 100% efficient. Unrepaired DNA lesions change gene expression, suppress cell division, or cause cell death [20-22]. DNA damage also plays a major role in mutagenesis and carcinogenesis. Tomasetti C and his colleagues showed that DNA replication errors contributed to two-thirds of the mutations found in 17 cancer types [23].
Evidence has shown that damage to both nuclear DNA (encodes most cellular RNA and proteins) and mitochondrial DNA is associated with aging [24].
Cellular DNA damage has also been shown to be involved in the aetiology and progression of many human diseases, such as Xeroderma pigmentosum, Ataxia-telangectasia, and Bloom's and Werner's syndromes.
Of course, mutations aren't meant to be permanent villains. Imagine if DNA repair were perfect enough to repair all DNA damage and no mutations ever built up, there would be no genetic changes, the raw material of evolution. Depending upon the conditions, DNA damage can be either causative or therapeutic.
DNA lesions can cause genetic alterations, and if genes that control cell growth are involved, these mutations can lead to the development of cancer. Damage to DNA may also lead to cell death which can have serious consequences for the organism of which the cell is part; for example, loss of irreplaceable neurons in the brain. Accumulation of damaged DNA has also been considered to contribute to some of the features of aging.
Although damage to human genomic DNA occurs very frequently, most of these lesions are successfully repaired by various repair mechanisms. If DNA lesions are not timely repaired or are too egregious to be repaired, there will trigger several signaling events that lead to three different cell fates, including senescence, apoptosis, or cancer transition.
DNA repair involves many pathways, some of which involve many proteins. To help research into DNA damage and repair, CUSABIO offers a number of related proteins.
References
[1] Sancar A., Lindsey-Boltz L.A., et al. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints [J]. Annu. Rev. Biochem. 2004; 73: 39-85.
[2] Lindahl T. Instability and decay of the primary structure of DNA [J]. Nature. 1993; 362: 709-715.
[3] Ganai R.A., Johansson E. DNA Replication—A Matter of Fidelity [J]. Mol. Cell. 2016;62:745–755.
[4] Redon CE, Nakamura AJ, et al. Histone gammaH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers [J]. Clin Cancer Res. 2010;16:4532–4542.
[5] Aparicio T, Baer R, Gautier J. DNA double-strand break repair pathway choice and cancer [J]. DNA Repair. 2014;19:169–175.
[6] Zhou BB, Elledge SJ 2000. The DNA damage response: Putting checkpoints in perspective [J]. Nature 408: 433–439.
[7] Matsuoka S, Huang M, Elledge SJ 1998. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase [J]. Science 282: 1893–1897.
[8] Reinhardt HC, Aslanian AS, et al. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage [J]. Cancer Cell 2007, 11: 175–189.
[9] Alexandre Maréchal and Lee Zou. DNA Damage Sensing by the ATM and ATR Kinases [J]. Cold Spring Harb Perspect Biol. 2013 Sep; 5(9): a012716.
[10] Lombard D.B., Chua K.F., et al. DNA repair, genome stability, and aging [J]. Cell. 2005; 120: 497-512.
[11] Hoeijmakers JHJ (2009). DNA damage, aging, and Cancer [J]. New England Journal of Medicine 361:1475–1485.
[12] Kunkel TA. DNA replication fidelity [J]. J Biol Chem. 2004;279(17):16895–16898.
[13] Kunkel TA. Evolving views of DNA replication (in)fidelity [J]. Cold Spring Harb Symp Quant Biol. 2009;74:91–101.
[14] Kunkel TA. Balancing eukaryotic replication asymmetry with replication fidelity [J]. Curr Opin Chem Biol. 2011;15(5):620–626.
[15] Ranjha, L., Howard, S. M. & Cejka, P. Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes [J]. Chromosoma 127, 187–214 (2018).
[16] Lodovichi, S. et al. Effect of BRCA1 missense variants on gene reversion in DNA double-strand break repair mutants and cell cycle-arrested cells of Saccharomyces cerevisiae [J]. Mutagenesis 35, 189–195 (2020).
[17] Roth, D. B. & Wilson, J. H. Relative rates of homologous and nonhomologous recombination in transfected DNA [J]. Proc. Natl Acad. Sci. USA 82, 3355–3359 (1985).
[18] Deckbar D, Jeggo PA, et al. Understanding the limitations of radiation-induced cell cycle checkpoints [J]. Crit Rev Biochem Mol Biol. 2011;46:271–283.
[19] Chang, H. H. Y., Pannunzio, N. R., Adachi, N., and Lieber, M. R. (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair [J]. Nat. Rev. Mol. Cell Biol. 18, 495–506.
[20] Zhou BBS, Elledge SJ. The DNA damage response: putting checkpoints in perspective [J]. Nature. 2000;408:433–439.
[21] Norbury CJ, Hickson ID. Cellular responses to DNA damage [J]. Ann Rev Pharmacol Toxicol. 2001;41:367–401.
[22] Kent S. Gates. An Overview of Chemical Processes That Damage Cellular DNA: Spontaneous Hydrolysis, Alkylation, and Reactions with Radicals [J]. Chem Res Toxicol. 2009 Nov; 22(11): 1747–1760.
[23] Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention [J]. Science. 2017;355(6331):1330–4.
[24] Karanjawala Z.E., Lieber M.R. DNA damage and aging [J]. Mech. Ageing Dev. 2004; 125: 405-416.


Comments
Leave a Comment