Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
| Code | CSB-EP007932HU |
| Abbreviation | Recombinant Human F8 protein |
| MSDS | |
| Size | $388 |
| Order now | |
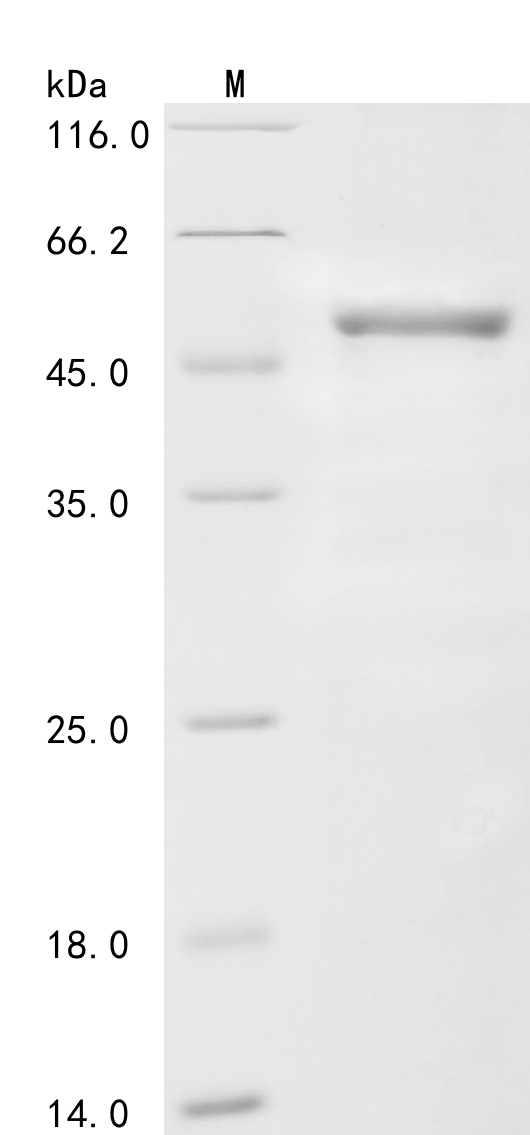
| Image |
|
| Have Questions? | Leave a Message or Start an on-line Chat |
To produce recombinant human Coagulation factor VIII (F8) in E. coli, the target gene fragment tagged with an N-terminal 6xHis-GST gene is cloned into an expression plasmid and transformed into E. coli cells. The target gene fragment encodes the 1-216aa of human Coagulation factor VIII. The cells are grown and induced for protein expression. The cells are lysed to release the recombinant protein, which is purified using affinity chromatography techniques. Protein purity is evaluated by SDS-PAGE, reaching up to 85%.
Human FVIII (F8) plays a crucial role in the blood coagulation cascade. It acts as a cofactor for the activation of factor X by factor IXa, thereby facilitating the formation of blood clots [1][2]. FVIII typically binds to the von Willebrand factor (vWF) in plasma, further enhancing its stability and function [3].
FVIII is a glycoprotein highly sensitive to proteolysis but is protected by forming a high-affinity complex with coagulation factor VIII-related antigen, which is subsequently released into the circulation [4]. It is vital for proper blood clotting [5]. FVIII constitutes only a small fraction of the proteins in plasma, typically ranging from 1 to 3 ppm [6]. Acquired hemophilia A, an autoimmune disease, can lead to life-threatening hemorrhagic disorders due to the presence of autoantibodies against FVIII [7][8].
References:
[1] G. Kemball‐Cook, E. Tuddenham, & A. Wacey, The factor viii structure and mutation resource site: hamsters version 4, Nucleic Acids Research, vol. 26, no. 1, p. 216-219, 1998. https://doi.org/10.1093/nar/26.1.216
[2] M. Kosloski, R. Miclea, & S. Balu‐Iyer, Role of glycosylation in conformational stability, activity, macromolecular interaction and immunogenicity of recombinant human factor viii, The Aaps Journal, vol. 11, no. 3, 2009. https://doi.org/10.1208/s12248-009-9119-y
[3] K. Pflegerl, A. Podgornik, E. Berger, & A. Jungbauer, Direct synthesis of peptides on convective interaction media monolithic columns for affinity chromatography, Journal of Combinatorial Chemistry, vol. 4, no. 1, p. 33-37, 2001. https://doi.org/10.1021/cc0100060
[4] H. Yamane, N. Ochi, T. Tabayashi, L. Lu, T. Yamagishi, Y. Monobeet al., Elevated coagulation factor viii plasma activity in a patient with lymphangiosarcoma, Internal Medicine, vol. 51, no. 22, p. 3213-3215, 2012. https://doi.org/10.2169/internalmedicine.51.8083
[5] C. Parmenter and S. Stoilova‐McPhie, Binding of recombinant human coagulation factor viii to lipid nanotubes, Febs Letters, vol. 582, no. 12, p. 1657-1660, 2008. https://doi.org/10.1016/j.febslet.2008.04.018
[6] O. Nordfang, M. Ezban, J. Favaloro, H. Dahl, & J. Hansen, Specificity of monoclonal antibodies to factor viii: c, Thrombosis and Haemostasis, vol. 54, no. 03, p. 586-590, 1985. https://doi.org/10.1055/s-0038-1660075
[7] Y. Otaki, R. Kouda, T. Fujimura, T. Nakatsue, M. Wakasugi, S. Murakamiet al., Acute renal failure as a complication of acquired hemophilia due to autoantibody to factor viii, Clinical and Experimental Nephrology, vol. 14, no. 1, p. 85-89, 2009. https://doi.org/10.1007/s10157-009-0226-y
[8] S. Kim, S. Park, C. Park, S. Choi, & J. Kim, Spontaneous retroperitoneal hemorrhage caused by idiopathic acquired hemophilia a misdiagnosed as a delayed traumatic hematoma: a case report, Journal of Acute Care Surgery, vol. 9, no. 2, p. 72-75, 2019. https://doi.org/10.17479/jacs.2019.9.2.72
There are currently no reviews for this product.

