Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Histone post-translational modifications (PTMs), also short for histone modifications, including methylation, acetylation, phosphorylation, etc. play an important role in various biological activities such as chromosome packaging, transcriptional regulation, and DNA damage and repair [1-4]. Therefore, accurate and sensitive detection of histone modifications is of great significance for understanding towards epigenetic regulation of biological processes and the development of histone-modifying enzymes (HME)-targeted drugs.
Histone modification detection plays an important role in revealing the key mechanisms of epigenetic regulation. By understanding the importance, principle, and detection methods of histone modification, we can better understand the key mechanisms of histone modification in gene regulation, development, and disease.
Histone modification assay is a series of experimental methods to analyze and determine the modification status and location of histones, mainly including chromatin immunoprecipitation (CHIP) and mass spectrometry (MS). These methods utilize specific experimental strategies and tools to detect histone molecules labeled with specific modifications and quantitatively determine the abundance and distribution of their presence.
ChIP is a commonly used histone modification assay technique to detect the presence of modifications and their association with gene expression. This technique uses specific antibodies to bind to modified histones to enrich the modified histones from cells or tissues. The enriched modified histones can then be further analyzed by PCR, sequencing, or mass spectrometry to determine the presence and localization of the modification.
There are three main CHIP-based methods for detecting histone modification, including ChIP-chip, ChIP-SAGE, and ChIP-Seq.
For ChIP, there are two types of ChIP: Native ChIP and Cross-linking ChIP. Native ChIP uses native chromatin prepared by nuclease digestion of cell nuclei. Cross-linking ChIP uses chromatin fixed with formaldehyde and fragmented by sonication. In table 1, we have a comparison between the two ways of ChIP.
Table 1. The Comparison Between Native ChIP And Cross-linking ChIP
| Types | advantages | disadvantages |
|---|---|---|
| Native ChIP |
|
|
| Cross-linking ChIP |
|
|
Besides, you can click the following name of protocols to view the entire protocols.
Native Chromatin immunoprecipitation (ChIP) Protocol
Cross-linking Chromatin immunoprecipitation (ChIP) Protocol
ChIP-chip (chromatin immunoprecipitation followed by microarray hybridization) is a technology that combines chromatin immunoprecipitation with DNA microarray. Like regular ChIP, ChIP-chip is used to investigate interactions between proteins and DNA in vivo. Actually, the ChIP-chip method can be used to study many of the epigenomic phenomena. The example that emerged here shows the application of ChIP-chip in studying histone modifications. As the Figure 1 shows:
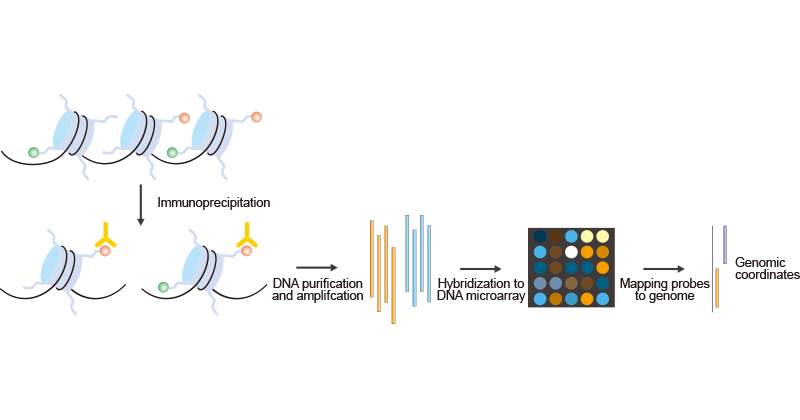
Figure 1. The Brief Processes of ChIP-on-chip
Modified chromatin is first purified by immunoprecipitating cross-linking chromatin with an antibody that is specific to a particular histone modification. Then DNA is amplified to obtain sufficient DNA (labeled with one color). The color-labeled ChIP DNA is hybridized into a DNA microarray with the control DNA prepared from input chromatin and labeled with a different color. Subsequently, the microarray probes can be mapped to the genome to yield genomic coordinates. However, most ChIP-on-chip experiments still use polyclonal antibodies, which are different in specificity between batches. The generation and use of monoclonal antibodies for ChIP-on-chip is therefore an important goal.
ChIP–SAGE refers to chromatin immunoprecipitation combined with serial analysis of gene expression. The combination of ChIP experiments with SAGE can profile histone modifications at a genomic scale. The ChIP-SAGE procedure begins with a ChIP step to purify chromatin regions that are associated with a specific histone modification, and proceeds as follows. It is shown in Figure 2:
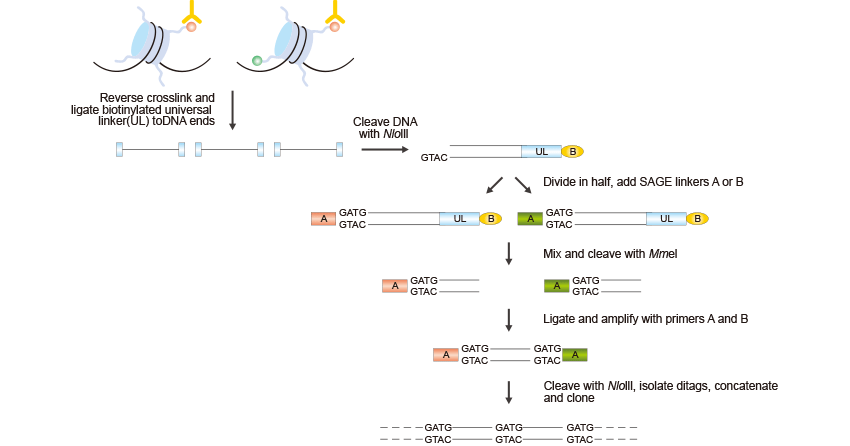
Figure 2. The Brief Processes of ChIP–SAGE
First of all, crosslinks are reversed, a biotinylated universal linker (UL) is ligated to DNA ends and DNA is bound to streptavidin beads. Then Nla III, which recognizes CATG, is used to digest DNA and a linker containing the recognition sequence of Mme I is ligated to the cleaved DNA ends. Mme I digestion produces 21-22 bp sequence tags from the immunoprecipitated fragments; the sequence tags are concatenated, cloned into a sequencing vector and sequenced. About 20 to 30 short sequence tags of 21 bp can be generated from each sequencing reaction. The sequence tags can then be mapped to the genome to identify modified regions.
ChIP-seq (chromatin immunoprecipitation combined with high-throughput sequencing techniques) is one of the most exciting recent advances in technologies for studying epigenetic phenomena at a genomic scale which relies on the combination of ChIP experiments with high-throughput sequencing. ChIP-seq combines ChIP with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. As the Figure 3 shows:
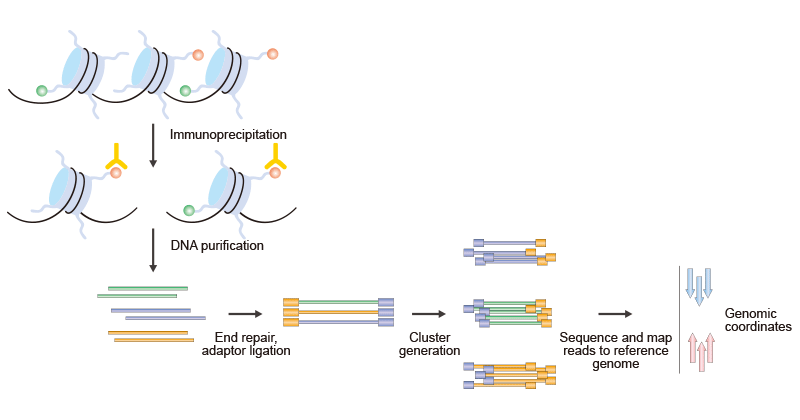
Figure 3. The Brief Processes of ChIP–Seq
The procedure that is outlined here is specific to the Illumina Genome Analyzer using Solexa technology. It can be divided into two parts, ChIP and Sequencing. The first step is the purification of modified chromatin by immunoprecipitation with an antibody that is specific to a particular histone modification. The ChIP DNA ends are repaired and ligated to a pair of adaptors, followed by limited PCR amplification. The DNA molecules are bound to the surface of a flow cell that contains covalently bound oligonucleotides that recognize the adaptor sequences. After size selection, all the resulting ChIP-DNA fragments are sequenced simultaneously using a genome sequencer. The resulting sequence reads are mapped to a reference genome to obtain genomic coordinates that correspond to the immunoprecipitated fragments.
Mass spectrometry has recently become the preferred method for the identification and quantification of histone modifications. Identification and specific residues’ localization of histone modifications through mass spectrometry rely on the detection of a "delta-mass" between the expected and experimentally measured masses of peptides or proteins. In theory, this approach enables the profiling of any PTM or combinations of PTMs in a single analysis in spite of the PTM type or its specific site, all while providing highly accurate quantitative data.
Mass spectrometry has proven to be a valuable tool for assessing and comparing the collective levels of particular modifications and their combinations across various samples.
There are three mass spectrometry-based methods that can be used to detect histone modifications in biological samples, including 'top-down', 'middle‐down' and 'bottom-up' mass spectrometry approaches [5-8].
In 'top-down' approach, intact histones are chromatographically isolated and subsequently ionized and subjected to mass spectrometry analysis [5,8]. High-resolution MS and MS/MS are essential for this technique due to the high charge state of intact histones, which can range from 13+ to 25+ for H3. This process yields comprehensive data on all histone isoforms within a given sample, along with their overall stoichiometry.
In 'middle‐down' approach, long histone peptides, typically exceeding 5 kDa in molecular weight (>20 amino acids), are generated through digestion with Glu‐C or Asp‐N [5,8]. Intact N-terminal tails can be obtained through this method. For instance, Asp-N produces the H4 1–24 peptide, while Glu-C yields the H3 1–50 peptide, which encompasses a majority of the well-known PTM sites found in histones H4 and H3, respectively.
The 'top-down' and 'middle-down' MS approaches face one of the major difficulties is to discriminating isobaric peptides (peptides bearing identical modifications but at distinct positions), although these two approaches are well-suited for detecting long-range PTM associations and estimating their abundance. Additionally, these approaches often grapple with reduced sensitivity and computationally intensive data analysis. Consequently, 'top-down' and 'middle-down' MS techniques are typically conducted in a limited number of specialized laboratories, and there have been no documented applications to clinical samples as of now.
In 'bottom-up' mass spectrometry method, histones undergo Arg‐C protease-mediated enzymatic digestion into relatively short peptides (5-20 amino acids in length) before being analyzed by mass spectrometry. Trypsin produces peptides that are too short due to core histones' rich basic amino acid residues, and inefficiently cleaves near modified sites, resulting in inconsistent lengths unsuitable for accurate quantification. Prior to trypsin cleavage, free amine derivatization on the N-terminal and lysine by deuterated acetic anhydride or propionic anhydride is often applied to simulate Arg-C digestion while using powerful trypsin [9-12]. This strategy also helps to distinguish isobaric peptides [10].
While 'bottom-up' provides limited co-occurring PTM data (up to four), especially for distant marks, it offers flexibility and has applications in analyzing patient-derived samples.
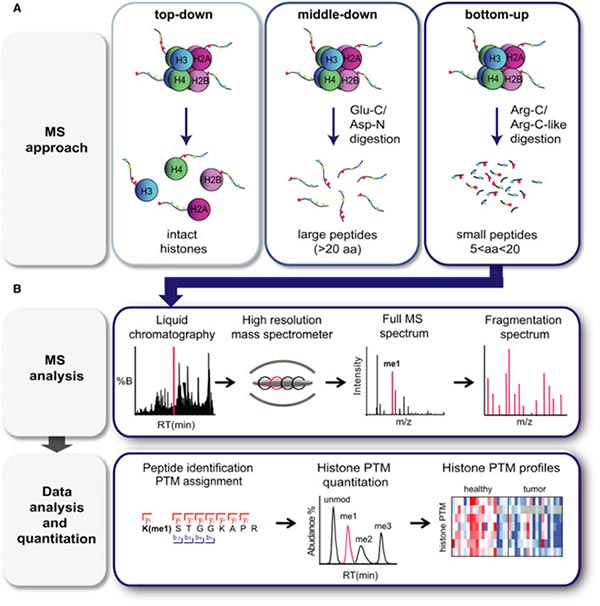
Figure 4. Mass spectrometry‐based analysis of histone PTMs.
The picture is cited from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9291046/
Histone modification detection is essential in epigenetics research and personalized medicine. It helps understand gene regulation, explore the process of cell differentiation and tissue development, identify disease biomarkers, and develop targeted therapies.
Histone modification detection can help us reveal the mechanism of gene regulation. By analyzing the distribution of different modifications in gene promoters and enhancers, we can understand the regulatory role of histone modifications in gene expression and further understand the detailed mechanism of gene regulation.
Histone modification detection plays an important role in the development process and cell fate determination. By analyzing the dynamic changes of modifications, we can study the molecular mechanisms of cell differentiation and tissue development, as well as the regulation of stem cell fate.
The detection of histone modification helps us to understand the mechanism of disease. Abnormal histone modification patterns in different diseases can provide important clues about the occurrence and development of diseases, thus providing new targets and strategies for disease diagnosis and treatment.
References
[1] Valensisi, C., Liao, J.L., Andrus, C. et al. cChIP-seq: a robust small-scale method for investigation of histone modifications [J]. BMC Genomics 16, 1083 (2015).
[2] Peterson CL, Laniel MA. Histones and histone modifications [J]. Curr Biol. 2004;14(14):R546–51.
[3] Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications [J]. Cell Res. 2011;21(3):381–95.
[4] Binder H, Steiner L, et al. Transcriptional regulation by histone modifications: towards a theory of chromatin re-organization during stem cell differentiation [J]. Phys Biol. 2013;10(2):026006.
[5] O’Geen H, Echipare L, Farnham PJ. Using ChIP-Seq technology to generate high-resolution profiles of histone modifications [J]. Methods Mol Biol. 2011;791:265–86.
[6] Noberini R, Robusti G, Bonaldi T. Mass spectrometry-based characterization of histones in clinical samples: applications, progress, and challenges [J]. FEBS J. 2022 Mar;289(5):1191-1213.
[7] S. Sidoli, B.A. Garcia. Characterization of individual histone posttranslational modifications and their combinatorial patterns by mass spectrometry-based proteomics strategies [J]. Methods Mol. Biol., 1528 (2017), pp. 121-148.
[8] Z.-F. Yuan, A.M. Arnaudo, B.A. Garcia. Mass spectrometric analysis of histone proteoforms [J]. Annu. Rev. Anal. Chem., 7 (2014), pp. 113-128.
[9] A. Moradian, A. Kalli, M.J. Sweredoski, S. Hess. The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications [J]. Proteomics, 14 (2014), pp. 489-497.
[10] Smith CM, Haimberger ZW, et al. (2002) Heritable chromatin structure: mapping "memory" in histones H3 and H4 [J]. Proc Natl Acad Sci USA 99 (Suppl 4), 16454–16461.
[11] Soldi M, Cuomo A & Bonaldi T (2014) Improved bottom‐up strategy to efficiently separate hypermodified histone peptides through ultra‐HPLC separation on a bench top Orbitrap instrument [J]. Proteomics 14, 2212–2225.
[12] Sidoli S, Bhanu NV, Karch KR, Wang X & Garcia BA (2016) Complete workflow for analysis of histone post‐translational modifications using bottom‐up mass spectrometry: from histone extraction to data analysis [J]. J Vis Exp 111, 54112.
Histone Modifications
Histone Auxiliary
Related Articles
Related Pathways