

Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
World Hemophilia Day is celebrated annually on April 17 to raise awareness of hemophilia and other bleeding disorders. Hemophilia was once considered a royal disease in Europe [1]. It took many years for hemophilia to be redefined. Hemophilia is a hereditary hemorrhagic disorder with abnormal coagulation function. Its typical features are dysfunction of active thrombin generation, prolonged coagulation time, and a lifelong tendency to bleed after minor trauma. Severe hemophilias can spontaneously bleed with no injury. So hemophiliacs are also called "glass men".
Hemophilia is a life-long disease. It often develops in childhood. Hemophilia is mainly inherited through the family, and some patients are caused by gene mutations. There is no cure for hemophilia at present. Without prompt treatment, bleeding episodes can become a life-threatening emergency. The current standard clinical treatment for hemophilia is recombinant coagulation factor replacement therapy [2] [3]. However, repeated venipuncture is required for life, which brings great pain and inconvenience to patients and heavy economic and psychological burdens to patients' families and society.
The most frequent types of hemophilia are hemophilia A (classic hemophilia) and hemophilia B (Christmas Disease) [4].
Hemophilia A, accounting for about 80 to 85 percent of all hemophilia, is caused by a deficiency or lack of clotting factor VIII (FVIII) [5]. Approximately 1 in 10,000 people suffer from hemophilia A. 70% of hemophilia A cases are parently inherited and 30% of cases are caused by mutations in the clotting factor VIII gene.
Hemophilia B is caused by missing or defective clotting factor IX (FIX). It accounts for 15%-20% of hemophilia. Hemophilia B is mainly caused by spontaneous gene mutations and exhibits little family inheritance. For both hemophilia A and hemophilia B, the less clotting factor your body generates, the worse your symptoms are.
The encoding genes for factors VIII and factor IX are present in the long arm of chromosome X. Both hemophilia A and B are inherited via an X-linked recessive pattern where 100% of females born from affected fathers will be carriers, and none of the males born will be affected. Female carrier mothers have a 50% chance of having affected males and a 50% chance of having carrier females. Due to its X-linked inheritance pattern, geographical areas with a higher frequency of consanguineous marriages like Egypt have a higher prevalence of the disease.
If the mother is a hemophilia carrier and the father is healthy, half of the baby boys will inherit hemophilia, and half of the baby girls will be carriers. If the father is a hemophiliac and the mother is healthy, all the baby girls will be carriers and all the baby boys are normal. A hemophiliac carrier female and a hemophiliac male will give birth to a 25% chance of hemophiliac boys, 25% chance of healthy boys, 25% chance of hemophiliac girl carriers, and 25% chance of hemophiliac girls. If both parents are hemophiliacs, all their offspring get sick. So hemophilia patients are almost entirely male, and women are extremely rare.
When a blood vessel is injured, three mechanisms operate locally at the injury site to control bleeding. After trauma, the injurious stimulation immediately causes local vasoconstriction and narrowing of the vessel, minimizing blood loss. Second, the subintimal tissues exposed by intimal injury can activate the blood clotting system and a mass of platelets, leading to platelets' aggregation at the site of the damage and the formation of a platelet plug, thus preventing the blood from flowing out of the vessel [6]. The blood-clotting system is poised to become engaged instantaneously in the formation of blood clots if tissue is injured. Furthermore, coagulation factors in the circulation are activated step by step (similar to the domino effect) to produce thrombin [7]. Under the action of thrombin, the soluble fibrinogen is transformed into fibrin monomer, which subsequently forms insoluble fibrin polymer interwoven into a net, trapping blood cells inside and eventually forming a blood clot.
Thrombocytosis is a process in which prothrombin is activated in a certain order and the blood changes from a fluid state to a gel state that cannot flow. It is an important part of physiological hemostasis. The basic physiological processes include prothrombin activator formation, thrombin formation, and fibrin formation. Here is a picture that plots the coagulation pathway below.
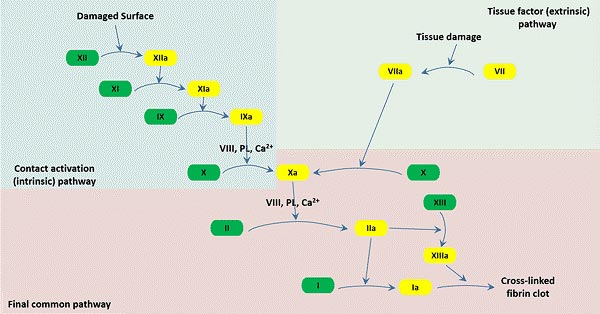
Figure 1: Three pathways that make up the classical blood coagulation pathway
In normal condition, 13 coagulation factors are involved in the coagulation process. However, in hemophilia patients, coagulation factors VIII and factor IX are lacking in the blood, interrupting the coagulation cascade, causing the body to fail to produce a sufficient amount of thrombin, ultimately leading to blood can not clot properly.
Coagulation factor is a general term for the substances directly involved in blood coagulation in plasma and tissues. At present, there are 12 types of coagulation factors numbered, namely coagulation factors I~XIII (VI is no longer regarded as an independent coagulation factor, it is actually the activated factor V). Except for factor IV, which is Ca2+, all the others are glycoproteins. The production of factors II, VII, IX, and X require the participation of vitamin K.
Table 1. The family of coagulation factors
| Name/Alias | Synthetic site | Involving pathway | Function |
|---|---|---|---|
| Factor I/Fibrinogen | Liver | Final common pathway | Forms fibrin |
| Factor II/Prothrombin | Liver | Final common pathway | Its active form (IIa) activates I, V, X, VII, VIII, XI, XIII, protein C, platelets |
| Factor III/Tissue factor |
Tissue Endotheliocyte monocyte |
Extrinsic pathway | Co-factor of VIIa |
| Factor IV/Calcium ion | --- | Three pathways | Required for coagulation factors to bind to phospholipid |
| Factor V/proaccelerin, labile factor | Liver | Final common pathway | Co-factor of X with which it forms the prothrombinase complex |
| Factor VII/stable factor, proconvertin | Liver | Extrinsic pathway | Activates IX, X |
| Factor VIII/Antihemophilic factor A | Liver | Intrinsic pathway | Co-factor of IX with which it forms the tenase complex |
| Factor IX/Antihemophilic factor B | Liver | Intrinsic pathway | Activates X: forms tenase complex with factor VIII |
| Factor X/Stuart-Prower factor | Liver | Final common pathway | Activates II: forms prothrombinase complex with factor V |
| Factor XI/plasma thromboplastin antecedent | Liver | Intrinsic pathway | Activates IX |
| Factor XII/Hageman factor | Liver | Intrinsic pathway | Activates factor XI, VII and prekallikrein |
| Factor XIII/fibrin-stabilizing factor | Liver, platelet | Final common pathway | Crosslinks fibrin |
If a newborn exhibits certain hemophilia symptoms, a preliminary screening test for coagulation function should be performed first. If coagulation function indicates a significantly prolonged APTT value, the newborn may have hemophilia. At this point, further clotting factor activity measurement is required. If the clotting factor activity measurement indicates an abnormally reduced clotting factor, the newborn is highly suspected of hemophilia. For patients with hemophilia, genetic testing for hemophilia is needed to further confirm the diagnosis and determine the type of hemophilia.
Screening tests are blood tests that can determine whether or not the blood is correctly clotting. Clotting factor tests reveal the type and severity of hemophilia.
Screening tests: prolonged prolonged activated partial thromboplastin time (APTT), bleeding time, prothrombin time, normal platelet
Confirmatory tests: FVIII activity assay, FIX activity assay, Ag (FVIII and FIX Antigen) assay
Genetic diagnosis: FVIII and FIX gene diagnosis
CUSABIO offers the following products for laboratory research on coagulation bleeding:
References
[1] Stevens RF. The history of haemophilia in the royal families of Europe [J]. Br J Haematol. 1999, 105: 25-32.
[2] Mannucci PM. Back to the future: a recent history of haemophilia treatment [J]. Haemophilia. 2008, 14 (Suppl. 3): 10-18.
[3] Mannucci PM: Hemophilia: treatment options on the twenty-first century. J Thromb Haemost. 2005, 1: 1349-1355.
[4] Bolton-Maggs PH, Pasi KJ. Hemophilias A and B [J]. Lancet. 2003, 361: 1801-1809.
[5] Hoyer LH. Hemophilia A [J]. N Engl J Med. 1994, 330: 38-47.
[6] Heemskerk JW, Bevers EM, Lindhout T. Platelet activation and blood coagulation [J]. Thromb Haemost. 2002;88:186–93.
[7] Furie B, Furie BC. Mechanisms of thrombus formation [J]. N Engl J Med 2008; 359:938.
Comments
Leave a Comment