

Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
"Don't be sad! The wheelchair is too tiny and I am just back to the universe". Stephen William Hawking said this on his deathbed. Hawking was diagnosed with amyotrophic Lateral Sclerosis in 1963 at 21 and was told that he had at most two years to live. However, he struggled and fought the disease for 55 years with solid volition and willingness to live. Hawking passed away for good on March 14, 2018. He created the wonder of both medical science and physics.
Most people know about ALS because Stephen Hawking, the great modern physicist, suffered from the disease. In 2014, celebrities such as Bill Gates of Microsoft, Mark Zuckerberg and Sandberg of Facebook, Jeff Bezos of Amazon, and Tim Cook of Apple, participated in the "Ice Bucket Challenge" letting the world know that there is such a group of people - amyotrophic lateral sclerosis.
Amyotrophic lateral sclerosis (ALS), also called Lou Gehrig's disease, was first identified and named by Jean-Martin Charcot, the father of French neurology, in 1869. ALS is a progressive neurological disease with irreversible muscle atrophy and paralysis, culminating in death from dysphagia and respiratory failure.
ALS mainly affects upper motor neurons (brain, brain stem, and spinal cord) and lower motor neurons (cranial nerve nucleus and spinal cord anterior horn cells), as well as its dominated torso and limbs and facial muscles. ALS has been listed by the WHO as one of the top five incurable diseases along with AIDS, cancer, leukemia, and rheumatoid disease.
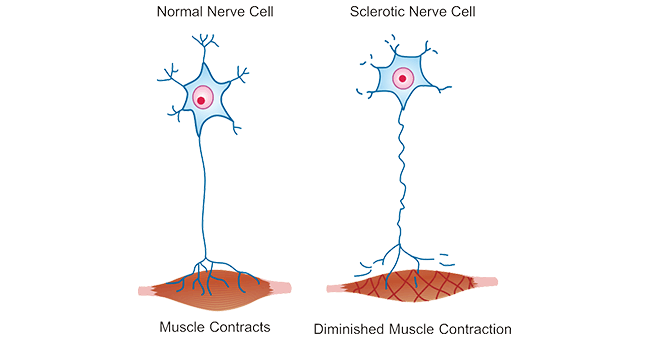
Figure 1. Comparison of normal nerve cell and sclerotic nerve cell
This picture is cited from: https://www.sciencedirect.com/science/article/pii/S2589958921000207
Like all neurodegenerative diseases, ALS is characterized by slow onset, progressive course, and poor prognosis. The course of the disease is rapid, only a few months to a few years. To make matters worse, it is very difficult to diagnose the disease in its early stages. It can take 9 to 15 months from the onset of symptoms to the diagnosis of ALS. The onset of ALS may be so subtle that the symptoms are overlooked.
The name ALS is a graphic description of the disease. People with ALS feel as if they are gradually frozen in ice, from their limbs to their torso and eventually to the muscles that control the movement of their eyes. Patients gradually lose the ability to walk and have difficulty swallowing and even breathing. ALS patients have even been called "conscious vegetative." Most ALS patients die from respiratory failure, usually within three to five years from when the symptoms first appear. Approximately 10% of patients survive for 10 or more years. About 90% of ALS cases are sporadic (SALS) and the remaining 10% are familial (FALS) [1].
There are many mechanisms and theories about the etiology of ALS, such as genetic factors, gene mutations, immunity, inflammation, specific food, and heavy metal poisoning. Most of the causes of ALS are unknown, genetic defects are involved in 20% of cases.
Among ALS patients, there is no exact epidemiological data in domestic at present. However, more and more people pay much attention to ALS due to the activity "Ice Bucket Challenge", which was very popular a few years ago. Although many ALS centers and laboratories worldwide have done a lot of research on the blood, cerebrospinal fluid, magnetic resonance imaging (MRI), and so on, they have not found an ALS biomarker with high sensitivity and specificity yet.
Researchers are making simulation studies on ALS potential biomarkers in many ways and from multi-angle. It has been found out from the patients with family history and with no family history that the LAS disease is associated with genetic mutations, including SOD1, TARDBP, FUS, UBQLN2, C9ORF72, etc. VAP protein may be involved in the pathogenesis of ALS, and other proteins such as CRP and Cystatin C are expected to become potential biomarkers.
In 1993, 11 different SOD1 missense mutations were identified in 13 different FALS families. Since then, more than 150 SOD1 mutations (including point mutations and protein truncation due to premature termination) have been identified in ALS patients, representing 20-25% of all FALS cases and a small fraction of SALS cases [2][3].
Importantly, ALS-associated SOD1 mutations induce protein misfolding and aggregation in axons, resulting in neuronal cell death. Thus, SOD1 has become one of the most widely studied genes in the field of ALS research and is considered a promising target for therapeutic intervention. Hawking suffered from ALS because of the SOD1 mutation.
Carsten Bonnemann and colleagues found that children with severe early-onset ALS had a rare mutation in the SPTLC1 gene, which encodes a key metabolic molecule responsible for the production of sphingolipids [4]. This report identified a novel metabolically related molecular pathway that may contribute to neurodegeneration in other subtypes of the disease.
Benjamin M. Neale group and Sali M. K. Farhan group of the Broad Institute of Harvard University and Massachusetts Institute of Technology, United States, have identified a new heat shock protein-encoding gene, DNAJC7, in the exome sequencing of ALS, and have revealed that DNAJC7 is a novel gene that regulates ALS [5].
ALS affects around 500,000 patients in the world, but there are still no effective treatment measures for this disease so far due to its unclear etiology. Treatments for ALS generally include gene therapy and cell transplantation, antioxidant and free radical scavenging, and new calcium channel blockers.
The U.S. Food and Drug Administration (FDA) approved Relyvrio (AMX0035) for the treatment of adult patients with ALS on local time September 29, 2022. AMX0035, developed by Amylyx Pharmaceuticals, is a combination of two drugs, Sodium Phenylbutrate and Taurursodiol, which can improve the health status of mitochondria and endoplasmic reticulum in cells, thereby delaying the death of nerve cells. It is the first treatment to significantly delay ALS progression and prolong survival in a randomized, placebo-controlled clinical trial and is only the third ALS treatment approved by the FDA in 116 years.
Prior to AMX0035's approval, only two drugs had been approved for ALS, Riluzole and Edaravone, but their effects were weak, in most cases delaying life by only a few months or slowing the process of deterioration. Although there are dozens of ALS drugs in development around the world, most do not stop or reverse the disease, or even substantially slow its progression.
Stem cell therapy has been used to treat ALS for decades. In recent years, the research of stem cell therapy for ALS has made good progress, prolonging the survival time of ALS patients, improving the neurological function of ALS patients, and curbing the progressive damage of motor neurons. Stem cell therapy would be an effective way to target ALS.
Although it is a huge challenge to explore therapies for ALS, many scientists have been never stopping grouping in the scientific road. Just as Hawking has set an example personally: "Don't let your physical inconveniences limit your soul!", don't let scientific research limit your imagination. Starting from basic research, CUSABIO manufactures and supplies all kinds of Elisa kits, which can help you target your scientific research and validation. We are glad to see that our products can work in your experiments, even if the effect is very tiny.
Here are some products related to Amyotrophic lateral sclerosis:
References
[1] A. C. Calvo et al., Amyotrophic lateral sclerosis: a focus on disease progression [J]. Biomed Res Int 2014, 925101 (2014).
[2] Rotunno MS and Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis [J]. Front Cell Neurosci. 2013 Dec 16;7:253.
[3] Prudencio M et al. An examination of wild-type SOD1 in modulating the toxicity and aggregation of ALS-associated mutant SOD1 [J]. Hum Mol Genet. 2010 Dec 15;19.(24):4774-89.
[4] Mohassel, P., Donkervoort, S., Lone, M.A. et al. Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis [J]. Nat Med 27, 1197–1204 (2021).
[5] Farhan, S.M.K., Howrigan, D.P., Abbott, L.E. et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein [J]. Nat Neurosci 22, 1966–1974 (2019).
Comments
Leave a Comment