Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
In immunology, glycobiology was once considered "dark matter"—complex and uncertain. However, as cancer immunotherapy hits a bottleneck, researchers realize the "glycocalyx" is not just a barrier but a sophisticated immune regulator. Specifically, the Siglec family decodes sialic acid signals and is emerging as a core immune checkpoint focus, following PD-1/PD-L1.
For researchers, the Siglec family offers both promise and challenges. With 15 members and complex ligand environments, determining research priorities is difficult. Success requires not only a deep understanding of structural biology but also a "research decision-making model" grounded in translational medicine logic.
The rise of the Siglec family is no accident; it parallels our deepening understanding of tumor immune evasion. In healthy tissues, surface sialylation acts as a "self" marker. Immune cells recognize these signals via inhibitory Siglecs to maintain tolerance and prevent autoimmunity. However, in the tumor microenvironment (TME), cancer cells induce "hypersialylation" through metabolic reprogramming. This creates a "sugar shield" that forcibly activates inhibitory Siglec signaling on immune cells, effectively extinguishing the immune attack [1].
This glycan-based suppression differs fundamentally from traditional protein-protein interactions like PD-1/PD-L1. It involves not just protein levels but also glycosylation enzyme kinetics, glycan conformations, and steric hindrance. Consequently, research has converged on a central theme: the Siglec family is the core hub of myeloid immunoregulation and a critical entry point for breaking tumor immune tolerance.
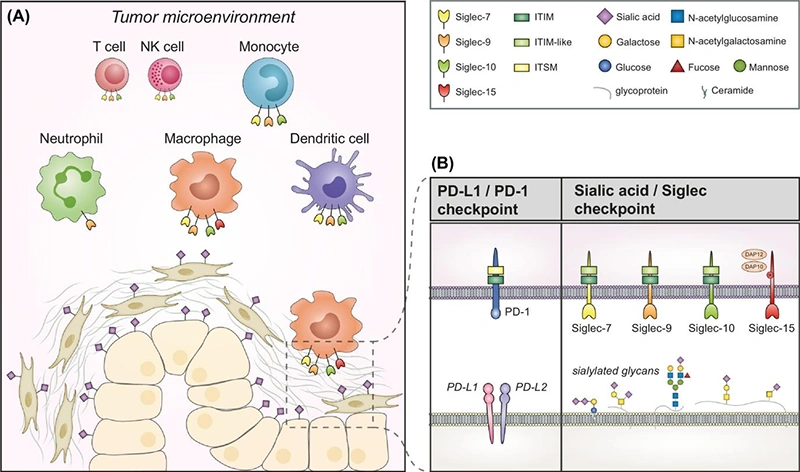
Figure: The Sialic Acid-Siglec Immune Checkpoint in the Tumor Microenvironment [2]
((A) Schematic representation of the expression of Siglec-7/9/10/15 and their sialic acid ligands in the TME. (B) A mechanistic comparison between the PD-1/PD-L1 checkpoint and the sialic acid-Siglec checkpoint.)
To conduct in-depth research on Siglecs, one must first understand their unique molecular architecture. Siglecs are type I transmembrane proteins and members of the immunoglobulin superfamily (IgSF). Their extracellular region is composed of a variable number of Ig-like domains. At the very end (the N-terminus) lies a characteristic V-set domain, which serves as the physical core for ligand recognition across all Siglec members.
Within this V-set domain, there is a highly conserved arginine residue. Spatially, this residue forms a positively charged "pocket" that creates a critical salt bridge with the carboxyl group of the sialic acid molecule. It is this precise, molecular-scale coupling that dictates the specific binding of Siglecs to sialic acid-containing glycans (sialoglycans) [3]. Following the V-set domain are a variable number of C2-set domains. These function like a "scaffold," pushing the recognition end out into the extracellular space. The number of these domains directly influences the receptor's "reach" and flexibility on the cell surface.

Figure: Schematic Representation of the Human Siglec Family Receptor Structures [4]
The human Siglec family comprises a total of 15 members, which are primarily divided into two subgroups based on their evolutionary and structural characteristics [5]:
These members are highly homologous across species and are responsible for fundamental physiological functions. For instance, with as many as 16 C2-set domains, Siglec-1 can perform long-range antigen capture on the surface of macrophages. Siglec-15 has also garnered significant attention due to its structural similarity to PD-L1 and its unique, bidirectional regulatory potential in tumor immunity.
This group evolves rapidly and is therefore also referred to as the fast-evolving type. They are primarily distributed on myeloid cells (neutrophils, monocytes, and macrophages). Their intracellular domains typically contain an immunoreceptor tyrosine-based inhibitory motif (ITIM) or an ITIM-like motif. Upon ligand binding, they recruit the phosphatases SHP-1/2 to transmit inhibitory signals, thereby exerting an "immune brake" function.
This group evolves rapidly and is therefore also referred to as the fast-evolving type. They are primarily distributed on myeloid cells (neutrophils, monocytes, and macrophages). Their intracellular domains typically contain an immunoreceptor tyrosine-based inhibitory motif (ITIM) or an ITIM-like motif. Upon ligand binding, they recruit the phosphatases SHP-1/2 to transmit inhibitory signals, thereby exerting an "immune brake" function.
It is noteworthy that certain members, such as Siglec-14 and Siglec-15, possess positively charged amino acids in their transmembrane regions. This allows them to interact with ITAM-containing adaptor proteins like DAP12, a feature typically associated with activating signals. However, high-throughput functional screening studies have revealed that Siglec-15, while dependent on DAP12 expression, paradoxically exhibits a sustained inhibitory effect on T-cell activity [6]. This complexity transcends traditional binary classifications of immune regulation.
The complexity of tumor immune evasion mechanisms lies in their multi-layered inhibitory network. Therefore, research decisions must start from a macro perspective to screen for Siglec targets with strategic complementarity. Current evidence indicates that the sialic acid-Siglec axis constructs a well-defined immunosuppressive system through the specific expression of different molecules on particular immune cell subsets. Based on this, research can focus on the following three core directions:
Addressing evasion strategies that impede effector cell function, Siglec-9 and Siglec-7 emerge as key research targets. Conclusive experimental evidence shows that highly sialylated mucins (e.g., MUC1) on the tumor cell surface act as ligands, specifically binding to Siglec-7 on NK cells and Siglec-9 on T cells and myeloid cells. This binding triggers an inhibitory signaling cascade mediated by their intracellular ITIM domains, directly leading to impaired killing activity in NK cells and hindered cytotoxic function in T cells [7]. Therefore, the core logic of this direction is to validate whether blocking this interaction can relieve the suppressed state of effector cells and restore the immune system's "immune surveillance" function against tumors.
Targeting innate immune evasion mechanisms, Siglec-10 demonstrates significant value as a core regulatory molecule. Extensive research confirms that the highly expressed CD24 on tumor cell surfaces acts as an "anti-phagocytic" signal. By binding to Siglec-10 on macrophages, it recruits and activates intracellular SHP-1/2 phosphatases, thereby transmitting a potent inhibitory signal that blocks macrophage phagocytosis [8]. This mechanism is particularly prominent in solid tumors such as breast and ovarian cancer. Consequently, incorporating Siglec-10 as a research target aims to explore the feasibility of reprogramming tumor-associated macrophages (TAMs) and restoring their phagocytic capacity by blocking the CD24-Siglec-10 axis.
Addressing compensatory and resistance mechanisms of immune checkpoints, Siglec-15 opens up a new track independent of the classic PD-1 axis. Key spatial transcriptomics data reveal that Siglec-15 is specifically highly expressed in various tumor tissues (e.g., non-small cell lung cancer, bladder cancer), and its spatial distribution shows significant mutual exclusivity with PD-L1. This implies that in PD-L1-negative "immune-cold tumors," Siglec-15 may independently mediate immune evasion through its unique characteristic of being DAP12 adaptor protein-dependent yet outputting inhibitory signals. Therefore, studying it as an independent direction helps to cover the PD-L1-negative patient population and elucidate the mechanisms of non-PD-1-dependent immunosuppressive remodeling.
Having clarified the differential positioning and strategic division of labor of the Siglec family within the tumor immune microenvironment, the logic of research must delve from macro-level target screening into the specific realm of intracellular signaling mechanisms. Myeloid cells, including TAMs and MDSCs, are the main executors of Siglec function. The signaling within these cells directly determines the efficacy of immune suppression. Therefore, the core of Step 2 is to decipher how Siglec receptors transmit inhibitory signals to reshape the functional states of myeloid cells.
Although the macro-level value of targets such as Siglec-9, -10, and -15 was established in Step 1, the key to therapeutic success lies in understanding the downstream processes. Specifically, how do the intracellular domains of these receptors, upon recognizing sialylated ligands on the myeloid cell surface, activate downstream signaling cascades via specific adaptor proteins (such as DAP12, SHP-1/2) to precisely regulate cell polarization, phagocytic function, or the secretion of inhibitory cytokines?
For instance, although Siglec-15 relies on the DAP12 adaptor protein, it exhibits inhibitory functions; how is this unique signal transduction feature utilized to achieve immune suppression within myeloid cells? Similarly, following the binding of Siglec-10 to CD24, how exactly does the activation of SHP-1/2 block the formation of the phagocytic synapse? These questions point to a deeper scientific inquiry: within the complex signaling network of myeloid cells, how does the Siglec axis, through molecular-level gaming, ultimately dictate the trajectory of the immune microenvironment?
Thus, the research decision for Step 2 should focus on this aspect, aiming to unveil the intracellular signal transduction mechanisms of myeloid cells and provide a direct theoretical basis for developing precise intervention strategies.
If your research is not confined to oncology but seeks to identify promising, differentiated targets within specific tissue contexts or pathological processes, the following members offer highly attractive entry points:
In the unique immune environment of the brain, the phagocytic capacity of microglia determines the clearance efficiency of amyloid-β (Aβ). High expression of CD33 on microglia has been proven to inhibit this phagocytosis, thereby increasing the risk of Alzheimer's disease (AD) [9]. In contrast, TREM2 provides an activating signal. The antagonistic balance between CD33 and TREM2 constitutes one of the most translationally promising research directions in current neuroimmunology.
Siglec-8 exhibits extreme tissue specificity—it is expressed exclusively on eosinophils, mast cells, and basophils. In diseases such as allergic asthma and chronic urticaria, activating Siglec-8 directly induces eosinophil apoptosis and inhibits mast cell degranulation [10]. Recent clinical studies have further confirmed that antibodies targeting Siglec-8 show significant efficacy in treating eosinophilic gastritis and duodenitis, effectively depleting eosinophils at gastrointestinal lesion sites [11]. This makes it one of the rare targets in inflammation research possessing both "tissue precision" and clinical translational potential.
CD22 is a negative regulator of B cell signaling; it modulates the activation threshold of the B cell receptor (BCR) by recognizing cis-distributed sialic acid ligands. In autoimmune diseases or B cell malignancies, CD22 serves not merely as a detection target but as the optimal model for studying receptor "masking effects" and ligand competition [12-13].
In Siglec research, the factor most likely to lead to biased conclusions is the neglect of its unique ligand biology. Addressing this is key to elevating Siglec research from "phenotypic description" to "mechanistic elucidation."
Most Siglecs are bound (masked) by sialic acid ligands on the cell surface in their resting state. Research confirms that these endogenous ligands occupy the receptors at extremely high local concentrations, preventing the binding of exogenous probes [14]. This implies that even if flow cytometry data indicates high receptor expression, the receptors may be functionally silent. When designing experiments, if sialidase is not used to remove this masking, the receptor binding capacity you observe may only be the tip of the iceberg.
Siglec recognition of sialic acid is highly fastidious; it not only distinguishes between α2,3- and α2,6- linkages but also depends heavily on the underlying glycan backbone and even the sequence context of the carrier protein. Recent studies confirm that sulfation modifications (such as those mediated by CHST1) and specific O-glycan patterns are key switches determining the binding of members like Siglec-3/7/8 [15]. Since the ligand preferences of CD22 and Siglec-8 differ drastically, relying solely on total sialic acid quantification or pan-specific probes can easily lead to biased conclusions; analysis must delve into single-site glycan structures and protein backgrounds.
A major hurdle in in vivo experiments is the rapid evolution of CD33-related Siglecs. Driven by host-pathogen selection pressure, these genes show significant divergence across species [16]. A key example is the human-specific loss of Neu5Gc, which fundamentally reshaped the ligand recognition spectrum of human Siglecs. Consequently, mouse homologs like Siglec-F or Siglec-E often fail to accurately simulate the biological functions of human Siglec-8 or Siglec-9. This discrepancy necessitates a cautious evaluation of the translational reliability of mouse model data.
Once you have determined the specific Siglec member and application scenario for your research, the next challenge is selecting experimental tools capable of supporting high-quality data. The choice of research tools should directly address your experimental questions:
If your question is "Can the receptor recognize a specific glycan ligand?": You require recombinant proteins with biological activity (such as Fc fusion proteins or conformationally stable His-tagged proteins). These tools are the cornerstone of SPR (Surface Plasmon Resonance) or ligand binding assays, enabling you to capture genuine binding signals rather than non-specific adsorption.
If your question is "Can blocking this axis reverse immune suppression?": In this case, high-specificity functional antibodies are core. You need to focus on whether the antibody can precisely target key binding sites within the V-set domain (such as the arginine pocket) and verify its neutralizing or activating capabilities in in vitro functional assays (e.g., T cell co-culture, macrophage phagocytosis assays).
If your question is "Quantitative analysis of the receptor in samples?": Then a high-sensitivity ELISA kit is the preferred choice. Kits that have undergone rigorous species-specific validation can precisely detect Siglec levels in serum, cell supernatants, or tissue lysates, providing you with reliable quantitative data and avoiding interference from cross-reactivity.
To help you precisely address the aforementioned scientific challenges, CUSABIO has carefully prepared a comprehensive Siglec research toolkit covering recombinant proteins, antibodies, and ELISA kits. This toolkit aims to provide you with one-stop support ranging from mechanism exploration to functional validation.
| Target | Code | Product Name | Source |
|---|---|---|---|
| CD22 | CSB-EP004900HU1 | Recombinant Human B-cell receptor CD22 (CD22), partial | E.coli |
| CD22 | CSB-MP004900HU | Recombinant Human B-cell receptor CD22 (CD22), partial (Active) | Mammalian cell |
| CD22 | CSB-MP004900HU-B | Recombinant Human B-cell receptor CD22 (CD22), partial, Biotinylated | Mammalian cell |
| CD33 | CSB-EP004925HU | Recombinant Human Myeloid cell surface antigen CD33 (CD33), partial | E.coli |
| CD33 | CSB-MP004925HU | Recombinant Human Myeloid cell surface antigen CD33 (CD33), partial (Active) | Mammalian cell |
| SIGLEC1 | CSB-EP021293PI | Recombinant Pig Sialoadhesin (SIGLEC1), partial | E.coli |
| SIGLEC9 | CSB-MP021303HU | Recombinant Human Sialic acid-binding Ig-like lectin 9 (SIGLEC9), partial (Active) | Mammalian cell |
| SIGLEC9 | CSB-MP5403MOV | Recombinant Macaca fascicularis (SIGLEC9), partial | Mammalian cell |
| SIGLEC10 | CSB-MP842706HU | Recombinant Human Sialic acid-binding Ig-like lectin 10 (Alpg), partial | Mammalian cell |
| SIGLEC15 | CSB-EP761623HU | Recombinant Human Sialic acid-binding Ig-like lectin 15 (SIGLEC15), partial | E.coli |
| SIGLEC15 | CSB-MP761623HU | Recombinant Human Sialic acid-binding Ig-like lectin 15 (SIGLEC15), partial (Active) | Mammalian cell |
| Target | Code | Product Name | Tested Applications |
|---|---|---|---|
| CD22 | CSB-RA962691A0HU | CD22 Recombinant Monoclonal Antibody | ELISA, IHC |
| CD22 | CSB-PA070294 | CD22 Antibody | WB, ELISA |
| CD22 | CSB-PA070293 | Phospho-CD22 (Y807) Antibody | WB, IHC, ELISA |
| CD22 | CSB-PA616669 | CD22 Antibody | ELISA, IHC |
| CD22 | CSB-PA576542 | CD22 Antibody | ELISA, IHC |
| CD22 | CSB-PA828492 | CD22 Antibody | ELISA, WB |
| CD22 | CSB-PA953915 | CD22 (Ab-807) Antibody | ELISA, WB |
| CD22 | CSB-PA912726 | Phospho-CD22 (Tyr807) Antibody | ELISA, WB |
| CD22 | CSB-PA004900ESR1HU | CD22 Antibody | ELISA, IHC |
| CD33 | CSB-PA006175 | CD33 Antibody | WB, ELISA |
| CD33 | CSB-PA004925GA01HU | CD33 Antibody | ELISA, WB, IHC, IF |
| CD33 | CSB-PA070366 | CD33 Antibody | ELISA, WB, IHC |
| CD33 | CSB-PA243594 | CD33 Antibody | ELISA, WB, IHC |
| CD33 | CSB-PA004925ESR1HU | CD33 Antibody | ELISA, WB, IHC |
| CD33 | CSB-PA004925ESR2HU | CD33 Antibody | ELISA, IHC |
| CD33 | CSB-PA004925LA01HU | CD33 Antibody | ELISA, WB, IHC |
| SIGLEC1 | CSB-PA005635 | SIGLEC1 Antibody | WB, ELISA |
| SIGLEC5 | CSB-PA005284 | SIGLEC5 Antibody | WB, ELISA |
| SIGLEC5 | CSB-PA021299GA01HU | SIGLEC5 Antibody | ELISA, WB |
| SIGLEC5 | CSB-PA571675 | SIGLEC5 Antibody | ELISA, IHC |
| Target | Code | Product Name | Detection Range | Sensitivity |
|---|---|---|---|---|
| CD33 | CSB-EL004925HU | Human Myeloid cell surface antigen CD33(CD33) ELISA kit | 125 pg/mL-8000 pg/mL | 31.25 pg/mL |
| SIGLEC8 | CSB-EL021302HU | Human Sialic acid-binding Ig-like lectin 8(SIGLEC8) ELISA kit | 0.156 ng/mL-10 ng/mL | 0.039 ng/mL |
| SIGLEC9 | CSB-EL021303HU | Human Sialic acid-binding Ig-like lectin 9(SIGLEC9) ELISA kit | 125 pg/ml-8000 pg/ml | 31.25 pg/ml |
[1] Feng H,Feng J,Han X, et al. The Potential of Siglecs and Sialic Acids as Biomarkers and Therapeutic Targets in Tumor Immunotherapy. Cancers (Basel). 2024;16 (2):.
[2] Boelaars K,van Kooyk Y. Targeting myeloid cells for cancer immunotherapy: Siglec-7/9/10/15 and their ligands. Trends Cancer. 2024;10 (3):230-241.
[3] Jame-Chenarboo Z,Gray TE,Macauley MS. Advances in understanding and exploiting Siglec-glycan interactions. Curr Opin Chem Biol. 2024;80:102454.
[4] Siddiqui SS,Matar R,Merheb M, et al. Siglecs in Brain Function and Neurological Disorders. Cells. 2019;8 (10):.
[5] Lin SY,Schmidt EN,Takahashi-Yamashiro K, et al. Roles for Siglec-glycan interactions in regulating immune cells. Semin Immunol. 2025;77:101925.
[6] Wang J,Sun J,Liu LN, et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med. 2019;25 (4):656-666.
[7] Rodriguez E,Boelaars K,Brown K, et al. Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat Commun. 2021;12 (1):1270.
[8] Barkal AA,Brewer RE,Markovic M, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572 (7769):392-396.
[9] Akinluyi ET,Akinluyi ET,Takahashi-Yamashiro K, et al. Interplay between CD33 and TREM2 in Alzheimer's Disease: Potential Mechanistic Insights into Microglial Function in Amyloid Pathology. ACS Chem Neurosci. 2026;17 (1):62-76.
[10] Kiwamoto T,Kawasaki N,Paulson JC, et al. Siglec-8 as a drugable target to treat eosinophil and mast cell-associated conditions. Pharmacol Ther. 2012;135 (3):327-36.
[11] Peterson KA,Chehade M,Genta RM, et al. Anti-Siglec-8 Antibody for Eosinophilic Gastritis and Duodenitis. N Engl J Med. 2020;383 (17):1624-1634.
[12] Frank MJ,Baird JH,Kramer AM, et al. CD22-directed CAR T-cell therapy for large B-cell lymphomas progressing after CD19-directed CAR T-cell therapy: a dose-finding phase 1 study. Lancet. 2024;404 (10450):353-363.
[13] Rhein S,Serin N,Timiliotis S, et al. A CD22-specific T-cell receptor enables effective adoptive T-cell therapy for B-cell malignancies. Blood. 2026;147 (10):1058-1069.
[14] Razi N,Varki A. Masking and unmasking of the sialic acid-binding lectin activity of CD22 (Siglec-2) on B lymphocytes. Proc Natl Acad Sci U S A. 1998;95 (13):7469-74.
[15] Büll C,Sun L,Van Coillie J, et al. Probing the binding specificities of human Siglecs by cell-based glycan arrays. Proc Natl Acad Sci U S A. 2021;118 (17):.
[16] Angata T,Varki A. Discovery, classification, evolution and diversity of Siglecs. Mol Aspects Med. 2023;90:101117.