Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Colony-stimulating factors (CSFs) are a family of secreted glycoproteins initially identified for their canonical ability to drive the proliferation, differentiation, and colony formation of hematopoietic progenitor cells in semisolid culture systems [1,2]. Over the past four decades, extensive research has expanded our understanding of CSFs beyond hematopoiesis, revealing their pivotal roles in immune homeostasis regulation, inflammatory response modulation, tissue repair, and tumor microenvironment (TME) remodeling.
This article provides a comprehensive overview of the classification, molecular characteristics, physiological functions, and canonical signaling pathways of core CSFs, with a particular focus on their translational and clinical applications in oncology, hematology, infectious diseases, and regenerative medicine. We further discuss the current challenges in CSF-based therapeutics and highlight emerging research frontiers, providing a systematic and up-to-date reference for researchers and scientists in biology and medicine.
Table of Contents
1. What Are Colony Stimulating Factors?
2. Core Family Members and Biological Functions of Colony Stimulating Factors
3. Canonical Signaling Pathways of Colony Stimulating Factors
4. Pathophysiological Roles and Therapeutic Targeting of CSF-Mediated Signaling
CSFs are a group of hematopoietic growth factors that bind to specific cell surface receptors on hematopoietic stem cells (HSCs) and committed progenitor cells, activating intracellular signaling cascades that drive cell proliferation, differentiation, survival, and functional activation of myeloid lineage cells, primarily granulocytes, monocytes, and macrophages [2-4].
Beyond their canonical role in steady-state and emergency hematopoiesis, accumulating evidence demonstrated that CSFs also play critical roles in innate and adaptive immunity, inflammation, tissue homeostasis, and cancer pathogenesis [2,5,6].
The mammalian CSF family consists of four well-characterized core members: granulocyte colony-stimulating factor (G-CSF, CSF3), granulocyte-macrophage colony-stimulating factor (GM-CSF, CSF2), macrophage colony-stimulating factor (M-CSF, CSF1), and multi-CSF (IL-3), each with non-redundant and partially overlapping biological activities, as well as related cytokines with CSF-like activity [1,2].
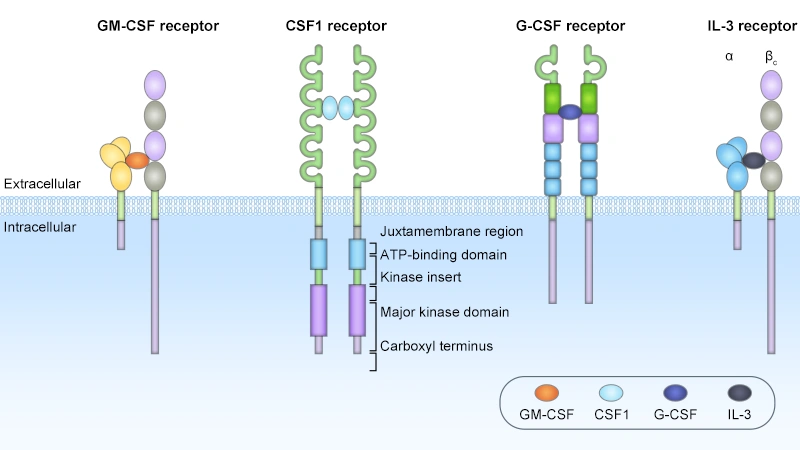
Figure. The structures of the CSF receptors
Comparison of Four Colony-Stimulating Factors (CSFs):
| CSFs | Key Features of CSFs | Receptors | Key Features of Receptor | Core Biological Functions & Ligand-Receptor Interaction Effects |
|---|---|---|---|---|
| G-CSF (CSF3) | The primary and most lineage-restricted regulator of neutrophil production in vivo [4,7]; Constitutively and inducibly produced by endothelial cells, monocytes, macrophages, and fibroblasts in response to inflammatory stimuli (LPS, TNF-α, IL-1β) [4,7] | G-CSFR (CSF3R) | Predominantly expressed on neutrophil lineage precursors and mature neutrophils [4,8] | Promotes survival, proliferation, and terminal differentiation of neutrophil progenitor cells in bone marrow; enhances phagocytic and bactericidal activity of mature neutrophils; potently induces mobilization of HSCs and hematopoietic progenitor cells from bone marrow into peripheral circulation [4,7,9]. |
| GM-CSF (CSF2) | Produced by activated T cells, macrophages, endothelial cells, and stromal cells; expression is tightly regulated during inflammatory and immune responses [10,11] | GM-CSFR (CSF2R): CSF2RA-βc | Heterodimeric complex composed of cytokine-specific α subunit (GM-CSFRα/CD116) and common β (βc) subunit shared with IL-3 and IL-5 receptors [11,15]. Expressed on myeloid progenitor cells, monocytes/macrophages, DCs, neutrophils, and some lymphocyte subsets | Exhibits a broader hematopoietic activity spectrum than G-CSF, regulating the development of granulocyte, macrophage, DC, eosinophil, and megakaryocyte lineages (at high concentrations) [2,10]. Beyond hematopoiesis, GM-CSF is a key regulator of innate and adaptive immunity: it promotes DC maturation and antigen presentation, enhances macrophage effector function, and drives pathological inflammation in autoimmune diseases [10,11]. |
| M-CSF (CSF1) | Constitutively expressed by most stromal cells, endothelial cells, and osteoblasts; upregulated during inflammation and tissue repair [12,17] | M-CSFR (CSF1R, CD115) | Class III receptor tyrosine kinase (RTK) with an extracellular domain containing five Ig-like loops for ligand binding, a transmembrane domain, and an intracellular tyrosine kinase domain [12,17]. Predominantly expressed on monocyte/macrophage lineage cells, osteoclasts, and CNS microglia | Principal regulator of monocyte-macrophage lineage, essential for development, survival, proliferation, and functional polarization of tissue-resident macrophages and circulating monocytes [2,12]. Critically involved in bone remodeling (via osteoclast development regulation), lipid metabolism, tissue repair, and modulation of tumor microenvironment through recruitment and polarization of tumor-associated macrophages (TAMs) [12,13]. |
| IL-3(Multi-CSF) | Primarily produced by activated CD4+ T cells; minor expression from mast cells and natural killer (NK) cells [15] | IL-3R: IL3RA-βc | Heterodimeric complex composed of ligand-specific α subunit (IL-3Rα/CD123) and shared βc subunit, forming a high-affinity complex similar to GM-CSFR [15]. IL-3Rα is highly expressed on leukemic stem cells in acute myeloid leukemia (AML) [1,20] | Acts on earlier, more primitive hematopoietic progenitor cells than other CSFs, supporting survival and proliferation of multipotent HSCs and development of multiple myeloid lineages (granulocytes, monocytes, megakaryocytes, erythroid cells) [1,2,15]. Not required for steady-state hematopoiesis, but plays a critical role in emergency hematopoiesis during infection and inflammation [15,18]. IL-3Rα is a promising therapeutic target for hematologic malignancies [1,20]. |
Despite differences in receptor structure and kinase activity, all CSF family members converge on four highly conserved canonical signaling cascades that mediate their core biological functions. These pathways act in a coordinated, often parallel manner to regulate survival, proliferation, differentiation, and effector functions of hematopoietic and myeloid cells.
The JAK/STAT pathway is the primary signaling cascade activated by G-CSF, GM-CSF, and IL-3, all of which signal through type I cytokine receptors that lack intrinsic kinase activity [14,16,20]. For G-CSF, receptor homodimerization brings associated JAK1 and JAK2 kinases into proximity, leading to their transphosphorylation and activation. Activated JAKs then phosphorylate tyrosine residues on the cytoplasmic domain of G-CSFR, creating docking sites for STAT3 and STAT5 proteins. Phosphorylated STAT proteins dimerize and translocate to the nucleus, where they regulate the transcription of target genes involved in cell survival, proliferation, and differentiation. For GM-CSF and IL-3, JAK2 is the primary kinase activated by the βc subunit, leading to the phosphorylation and activation of STAT5, as well as STAT3 in certain cell types [4,20].
The MAPK/ERK pathway is another core cascade activated by all CSFs. For G-CSF, activated JAKs phosphorylate adaptor proteins including Shc and Grb2, which recruit the guanine nucleotide exchange factor SOS to the receptor complex, leading to the activation of Ras GTPase. Activated Ras then initiates a phosphorylation cascade involving Raf, MEK, and extracellular signal-regulated kinase (ERK1/ERK2). Phosphorylated ERK translocates to the nucleus and regulates the transcription of genes associated with cell cycle progression, proliferation, and differentiation. For CSF-1, activated CSF-1R RTK directly phosphorylates adaptor proteins, including Shc and Grb2, activating the Ras/MAPK cascade to drive macrophage proliferation and survival [12,20].
All CSFs activate the PI3K/Akt pathway, which primarily mediates cell survival and anti-apoptotic effects. For G-CSF, activated JAKs and receptor phosphotyrosines recruit the p85 regulatory subunit of PI3K, leading to the activation of the p110 catalytic subunit and the production of PIP3. PIP3 recruits Akt to the plasma membrane, where it is phosphorylated and activated by PDK1 and mTORC2. Activated Akt phosphorylates multiple downstream targets, including Bad, caspase-9, and FOXO1 transcription factors, inhibiting apoptosis and promoting cell survival. For CSF-1, PI3K/Akt activation is critical for macrophage migration, cytoskeletal rearrangement, and functional polarization [12,20].
Colony Stimulating Factors are central regulators of myeloid cell biology, with signaling pathways that govern hematopoiesis, immunity, tissue homeostasis, and disease. However, dysregulation of CSF-mediated signaling drives a wide range of diseases, including chronic inflammation, autoimmunity, cancer, and neurodegeneration, making these pathways attractive therapeutic targets.
Dysregulated CSF signaling is a key driver of chronic inflammatory and autoimmune diseases. GM-CSF is a central mediator of autoimmune inflammation in rheumatoid arthritis, multiple sclerosis, and inflammatory bowel disease: elevated GM-CSF levels drive the accumulation and activation of pro-inflammatory macrophages and Th17 cells, promoting tissue damage [10,21]. Therapeutic monoclonal antibodies targeting GM-CSF or GM-CSFR are approved for the treatment of rheumatoid arthritis and are in clinical trials for multiple sclerosis and other autoimmune diseases [10,21].
CSF-1/CSF-1R signaling is implicated in chronic inflammatory diseases, including atherosclerosis, inflammatory arthritis, and neurodegenerative diseases. In atherosclerosis, CSF-1 drives macrophage proliferation and foam cell formation in atherosclerotic plaques, promoting disease progression [22]. In neurodegenerative diseases, including Alzheimer's disease and amyotrophic lateral sclerosis (ALS), dysregulated CSF-1R signaling in microglia drives chronic neuroinflammation and neuronal damage [2,23]. Small-molecule inhibitors of CSF-1R are in preclinical and clinical development for these diseases [2,23].
CSF-mediated signaling plays dual roles in cancer, with both tumor-suppressive and tumor-promoting effects depending on the context [19]. G-CSF is widely used clinically to reduce chemotherapy-induced neutropenia and febrile neutropenia in cancer patients, improving the safety and delivery of myelosuppressive chemotherapy [6,24].
However, aberrant CSF signaling also drives tumor progression and metastasis: CSF-1 is secreted by many solid tumors, recruiting tumor-associated macrophages (TAMs) to the tumor microenvironment (TME), where TAMs promote angiogenesis, extracellular matrix remodeling, immune suppression, and metastasis [23,25]. High CSF-1 and CSF-1R expression in tumors correlates with poor prognosis in breast, ovarian, pancreatic, and colorectal cancers [23,25]. Small-molecule CSF-1R inhibitors and anti-CSF-1R monoclonal antibodies are in clinical trials for multiple solid and hematologic malignancies, both as monotherapies and in combination with immune checkpoint inhibitors [23,25].
Activating mutations in the CSF3R gene are the defining genetic lesion in chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML), driving constitutive G-CSFR signaling and excessive neutrophil proliferation [26]. JAK inhibitors, including ruxolitinib, are effective in treating patients with CSF3R-mutant myeloid malignancies [26]. G-CSFR is also upregulated in many solid tumors, including breast, lung, and colorectal cancers, where G-CSF signaling drives tumor cell proliferation, survival, and metastasis, and promotes immunosuppression in the TME [6,24].
CSF-mediated signaling is critical for host defense against bacterial, fungal, and viral infections. G-CSF is used clinically to enhance neutrophil production in patients with severe congenital neutropenia, reducing the incidence of life-threatening bacterial infections [24,26]. GM-CSF has been investigated as an adjuvant for antiviral vaccines, including for COVID-19, due to its ability to enhance DC function and antigen-specific T cell responses [21]. GM-CSF also promotes the clearance of pulmonary pathogens and has been used to treat secondary bacterial pneumonia in critically ill patients [21].
Despite the tremendous success of CSF-based therapeutics, several key challenges remain, and ongoing research is addressing these limitations while exploring new frontiers of CSF biology and therapeutic applications.
One of the primary challenges is managing adverse effects associated with CSF administration. G-CSF is commonly associated with mild to moderate bone pain, headache, and fatigue, with rare but serious adverse events including splenomegaly, splenic rupture, and acute respiratory distress syndrome (ARDS). GM-CSF has a more pronounced pro-inflammatory effect, with common adverse events including fever, myalgia, capillary leak syndrome, and injection site reactions, which limit its systemic administration at high doses. CSF-1R inhibitors are associated with adverse effects, including fatigue, edema, elevated liver enzymes, and hematologic toxicities, as well as off-target effects on normal tissue macrophages [4,12,20].
Another major challenge is the development of resistance to CSF-targeted therapies, particularly CSF-1R inhibitors, in cancer treatment. Preclinical studies have identified multiple mechanisms of resistance, including compensatory upregulation of alternative chemokine pathways (e.g., CCL2/CCR2), activation of the PI3K/Akt/mTOR pathway, and epigenetic reprogramming of TAMs. In addition, the high cost of original CSF biologics has limited patient access in low- and middle-income countries, although the introduction of biosimilars has partially addressed this issue [12,27].
The tissue-specific delivery of CSFs also remains a challenge, as systemic administration can lead to off-target effects and limited exposure at the disease site. For example, systemic GM-CSF administration can promote systemic inflammation, while local delivery to the tumor or vaccination site is required for optimal anti-tumor immunity. For neurological indications, the blood-brain barrier (BBB) limits the penetration of systemically administered CSFs into the CNS, requiring the development of BBB-penetrant formulations or local delivery strategies [2,4].
The development of next-generation CSF biologics is a key research focus, with efforts to improve pharmacokinetic properties, reduce adverse effects, and enhance therapeutic efficacy. Novel long-acting G-CSF formulations with extended half-life allow for less frequent administration, improving patient adherence and quality of life [4]. Site-specific PEGylation and albumin fusion technologies are being used to optimize the half-life and bioactivity of CSFs, while reducing immunogenicity. In addition, bispecific molecules targeting CSFs and disease-specific antigens are being developed to enable targeted delivery, enhancing local therapeutic effects while minimizing systemic adverse events [4].
Biosimilars of CSFs will continue to play a critical role in improving global patient access, with ongoing clinical development and regulatory approval of new biosimilar products for G-CSF, GM-CSF, and other biologics. Real-world data from biosimilar use is accumulating, confirming their long-term efficacy and safety, and supporting their widespread adoption in clinical practice [27].
Combination strategies are being extensively explored to maximize the therapeutic efficacy of CSFs and CSF-targeted agents. In cancer immunotherapy, the combination of CSF-1R inhibitors with ICIs, chemotherapy, radiotherapy, and targeted therapies is being evaluated in preclinical and clinical studies, with the goal of reversing the immunosuppressive TME and overcoming ICI resistance [12]. Preclinical studies have shown that the triple combination of CSFs, radiotherapy, and ICIs induces synergistic anti-tumor immunity by promoting immunogenic cell death, enhancing antigen presentation, and depleting immunosuppressive TAMs [12,28].
In infectious diseases, the combination of CSFs with antibiotics and antiviral agents is being explored to enhance host immune defense against drug-resistant pathogens, particularly in immunocompromised patients. In regenerative medicine, the combination of CSFs with stem cell therapy and other growth factors is being evaluated to enhance tissue repair and regeneration in neurological and cardiovascular diseases [2,28].
Ongoing research is expanding the therapeutic indications of CSFs into new disease areas, including neurodegenerative diseases, autoimmune disorders, and metabolic diseases. Preclinical studies have identified a key role of the M-CSF/CSF-1R axis in the pathogenesis of Alzheimer's disease, Parkinson's disease, and multiple sclerosis, with CSF-1R inhibitors showing efficacy in reducing neuroinflammation and neurodegeneration in preclinical models [2,29]. Clinical trials are underway to evaluate CSF-targeted therapies in these neurodegenerative diseases to modify disease progression [2].
In addition, single-cell RNA sequencing and spatial transcriptomics technologies are revealing novel cellular sources and targets of CSFs in health and disease, uncovering previously unrecognized mechanisms of CSF action. For example, recent studies have identified a key role of CSFs in the regulation of tissue-resident macrophage populations in multiple organs, including the liver, lung, and adipose tissue, with implications for metabolic diseases and tissue fibrosis. These advances in basic research will continue to drive the development of novel CSF-targeted therapeutics for a wide range of human diseases [12,20,29].
CSFs are a family of secreted glycoproteins that regulate myeloid cell development, immune homeostasis, inflammation, tissue repair, and tumor microenvironment remodeling. Research on CSFs has not only deepened our fundamental understanding of hematopoiesis and immune biology but also led to transformative clinical applications in oncology, hematology, and infectious diseases, while offering promising therapeutic targets for autoimmune disorders, neurodegenerative diseases, and regenerative medicine.
References
[1] Metcalf D. The colony-stimulating factors and cancer [J]. Cancer Immunol Res. 2013 Dec;1(6):351-6.
[2] Chitu, V., Biundo, F., & Stanley, E. (2021). Colony Stimulating Factors in the Nervous System [J]. Seminars in Immunology, 54, 101511.
[3] Glaspy JA, Golde DW. The colony-stimulating factors: biology and clinical use [J]. Oncology (Williston Park). 1990 Sep;4(9):25-32; discussion 32-4.
[4] Link, H. (2022). Current state and future opportunities in granulocyte colony-stimulating factor (G-CSF) [J]. Supportive Care in Cancer, 30(9), 7067.
[5] Metcalf, D. (1989). The molecular control of cell division, differentiation commitment and maturation in haemopoietic cells [J]. Nature, 339(6219), 27-30.
[6] Hamilton, J. A. (2008). Colony-stimulating factors in inflammation and autoimmunity [J]. Nature Reviews Immunology, 8(7), 533-544.
[7] Dale, D. C. (1998). The discovery, development and clinical applications of granulocyte colony-stimulating factor [J]. Transactions of the American Clinical and Climatological Association, 109, 27.
[8] Gearing, D. P., King, J. A., Gough, N. M., & Nicola, N. A. (1989). Expression cloning of a receptor for human granulocyte-macrophage colony-stimulating factor [J]. The EMBO Journal, 8(12), 3667.
[9] Smith TJ, Khatcheressian J, et al. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline [J]. J Clin Oncol. 2006 Jul 1;24(19):3187-205.
[10] Hamilton, J. A. (2019). GM-CSF in inflammation [J]. The Journal of Experimental Medicine, 217(1), e20190945.
[11] Bagley, C. J., Woodcock, J. M., Stomski, F. C., & Lopez, A. F. (1997). The Structural and Functional Basis of Cytokine Receptor Activation: Lessons From the Common β Subunit of the Granulocyte-Macrophage Colony-Stimulating Factor, Interleukin-3 (IL-3), and IL-5 Receptors [J]. Blood, 89(5), 1471-1482.
[12] Yi, L., Gai, Y., et al. (2024). Macrophage colony-stimulating factor and its role in the tumor microenvironment: Novel therapeutic avenues and mechanistic insights [J]. Frontiers in Oncology, 14, 1358750.
[13] Cannarile, M. A., Weisser, M., et al. (2017). Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy [J]. Journal for Immunotherapy of Cancer, 5, 53.
[14] Schwartz, D. M., Villarino, A. V., et al. (2015). The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention [J]. Annual Review of Medicine, 66, 311.
[15] Miyajima A, Kinoshita T, et al. Signal transduction by the GM-CSF, IL-3 and IL-5 receptors [J]. Leukemia. 1997 Apr;11 Suppl 3:418-22.
[16] Rane SG, Reddy EP. JAKs, STATs and Src kinases in hematopoiesis [J]. Oncogene. 2002 May 13;21(21):3334-58.
[17] Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y, Yeung YG. Biology and action of colony--stimulating factor-1 [J]. Mol Reprod Dev. 1997 Jan;46(1):4-10.
[18] Ihle JN, Keller J, et al. Biologic properties of homogeneous interleukin 3. I. Demonstration of WEHI-3 growth factor activity, mast cell growth factor activity, p cell-stimulating factor activity, colony-stimulating factor activity, and histamine-producing cell-stimulating factor activity [J]. J Immunol. 1983 Jul;131(1):282-7.
[19] Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer [J]. Nat Rev Drug Discov. 2018 Dec;17(12):887-904.
[20] Barreda DR, Hanington PC, Belosevic M. Regulation of myeloid development and function by colony stimulating factors [J]. Dev Comp Immunol. 2004 May 3;28(5):509-54.
[21] Petrina, M., Martin, J., & Basta, S. (2021). Granulocyte macrophage colony-stimulating factor has come of age: From a vaccine adjuvant to antiviral immunotherapy [J]. Cytokine & Growth Factor Reviews, 59, 101-110.
[22] Sinha, S. K., Miikeda, A., et al. (2020). Local macrophage colony-stimulating factor expression regulates macrophage proliferation and apoptosis in atherosclerosis [J]. Arteriosclerosis, Thrombosis, and Vascular Biology, 41(1), 220.
[23] Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells [J]. Cold Spring Harb Perspect Biol. 2014 Jun 2;6(6):a021857.
[24] Park SD, Saunders AS, Reidy MA, Bender DE, Clifton S, Morris KT. A review of granulocyte colony-stimulating factor receptor signaling and regulation with implications for cancer [J]. Front Oncol. 2022 Aug 11;12:932608.
[25] Chen X, Liu H, Focia PJ, Shim AH, He X. Structure of macrophage colony stimulating factor bound to FMS: diverse signaling assemblies of class III receptor tyrosine kinases [J]. Proc Natl Acad Sci U S A. 2008 Nov 25;105(47):18267-72.
[26] Zhao X, Kawano S-i, Masuda J, Murakami H. G-CSF-dependent neutrophil differentiation requires downregulation of MAPK activities through Gab2 signaling pathway [J]. Cell Biol Int. 2020;44:1919–1933.
[27] Aapro, M. S., Chaplin, S., et al. (2023). Cost-effectiveness of granulocyte colony-stimulating factors (G-CSFs) for the prevention of febrile neutropenia (FN) in patients with cancer [J]. Supportive Care in Cancer, 31(10), 581.
[28] Benna, M., Guy, J. B., et al. (2020). Chemoradiation and granulocyte-colony or granulocyte macrophage-colony stimulating factors (G-CSF or GM-CSF): Time to think out of the box [J]? The British Journal of Radiology, 93(1109), 20190147.
[29] Stanley, E. R., Biundo, F., Gökhan, Ş., & Chitu, V. (2023). Differential regulation of microglial states by colony stimulating factors [J]. Frontiers in Cellular Neuroscience, 17, 1275935.


Comments
Leave a Comment