Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Emily Whitehead, diagnosed with relapsed acute lymphoblastic leukemia (ALL), was the first child to receive CAR-T therapy in 2012 after all standard treatments failed. The CAR-T cell therapy worked on Emily, and she is cancer-free today over a decade later. Emily’s story is a testament to the groundbreaking advancements in CAR-T cell therapy and offers new hope to cancer patients who have exhausted other treatment options.
In the relentless battle against cancer, the emergence of CAR-T cell therapy has marked a revolutionary leap in personalized medicine. Its journey from experimental innovation to clinical reality is a testament to the relentless pursuit of science and the promise of tailored therapies. This article delves into the pioneering advancements of CAR-T cell therapy, exploring its mechanisms, successes, and the challenges that lie ahead in the quest to redefine cancer treatment.
Table of Contents
1. Advances in CAR T-Cell Therapy Research
2. Key Parameters in CAR Design
The cancer cell therapy field is expanding rapidly, particularly in CAR-T cell research over the past three years (2020-2022) [1]. However, most therapies are still in early development stages, with a broad range of cell sources being explored. The CAR-T cell therapy industry shows strong momentum but is still largely pre-commercial, indicating both high potential and ongoing scientific challenges.
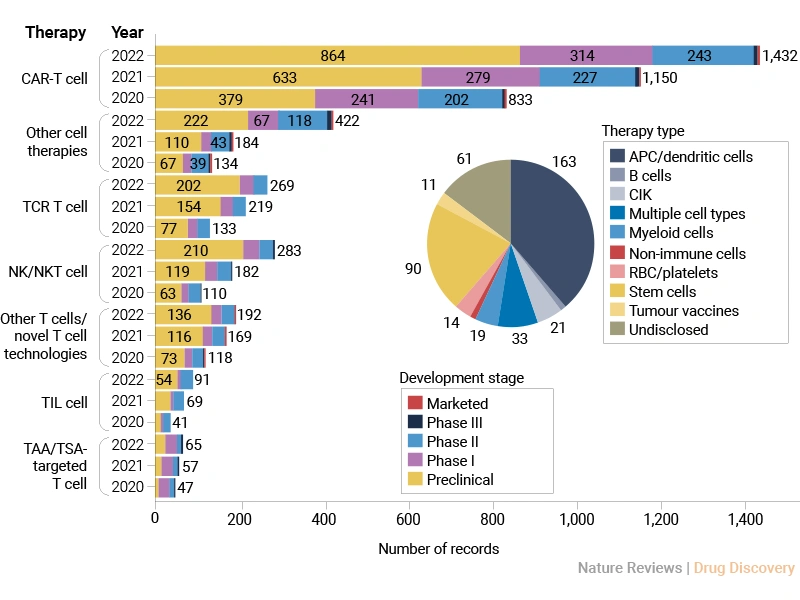
Figure 1. Changes in the cancer cell therapy pipeline by therapy type and year [1]
Chimeric antigen receptor T (CAR-T) cell therapy is a type of immunotherapy that harnesses the power of our immune system to fight cancer.
CAR-T cell therapy involves collecting blood from the patient to harvest T-cells, which are modified to express a CAR that recognizes and binds to specific tumor antigens. These engineered T cells are multiplied in the lab and infused back into the patient, where they identify the cancer cells with target antigens and kill them.
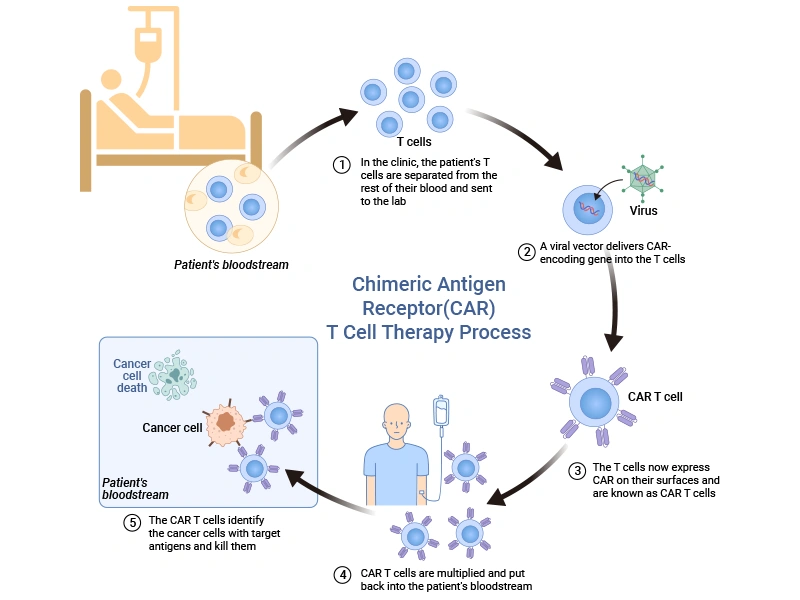
Figure 2. Schematic representation of the main process of CAR-T cell therapy [2]
CAR-T cell therapy has evolved over the decades. Scientists first proposed the idea of using T cells with synthetic receptors in the late 1980s. But it wasn’t until the last 15 years that technology allowed researchers to make it happen safely.
In 1987, the concept of double-chain chimeric T-cell receptor (cTCR) was proposed by Yoshikazu Kurosawa [3], followed by the first description of the first generation of CARs with scFv in 1993 [4]. Key co-stimulatory domains CD28 and 4-1BB were incorporated into T cells through the CAR construct in 2002 and 2004, respectively [5,6]. The first CAR-T trials in patients began in 2006 [7,8]. The third generation, containing two or more costimulatory domains, emerged in 2010, and the fourth generation was developed in 2012.
By 2011, leukemia patients achieved complete remission with second-generation CAR-T. In 2012, Emily Whitehead became the first pediatric patient treated with CAR-T. The FDA approved CD19 CAR-T cell therapy (Kymriah and Yescarta) for leukemia in 2017. In 2021, Abecma was approved for multiple myeloma [9].
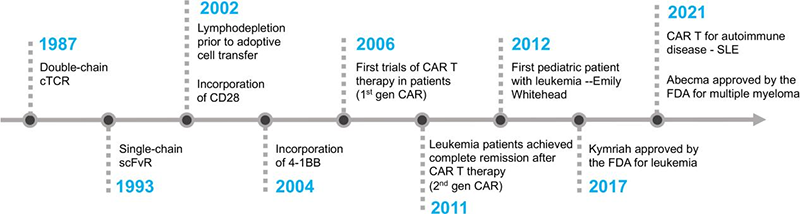
Figure 3. The timeline of key milestones in the development of CAR T cell therapy [10]
In 2017, the FDA approved the first CAR T-cell therapy for children and young adults with acute lymphoblastic leukemia (ALL). Soon after, approvals expanded to aggressive lymphomas and multiple myeloma.
Table 1. FDA-approved CAR-T cell therapies for B cell malignancies and multiple myeloma [10]
| Product Name | Brand Name | Company | Target | Approval Year | Diseases Approved for |
|---|---|---|---|---|---|
| Tisagenlecleucel | Kymriah | Novartis | CD19 | 2017 | R/R cell All, R/R DLBCL |
| Axicabtagene ciloleucel | Yescarta | Kite | CD19 | 2017 | R/R DLBCL,R/R FL |
| Brexucabtagene autoleucel | Tecartus | Kite | CD19 | 2020 | R/R MCL |
| Lisocabtagene maraleucel | Breyanzi | Juno/BMS | CD19 | 2021 | R/R LBCL |
| Idecabtagene vicleucel | Abecma | BMS | ВCMA | 2021 | R/R MM |
| Ciltacabtagene autoleucel | Carvykti | Legend/J&J | BCMA | 2022 | R/R MM |
| Obecabtagene autoleucel | Aucatzyl | Autolus | CD19 | 2024 | R/R B-ALL |
(Note: R/R, relapsed or refractory; ALL, Acute Lymphoblastic Leukemia; DLBCL, Diffuse Large B-cell Lymphoma; FL, follicular lymphoma; MCL, Mantle Cell Lymphoma; LBCL, Diffuse Large B-cell Lymphoma; MM, Multiple Myeloma; B-ALL, B-cell Acute Lymphoblastic Leukemia)
However, CAR-T is more than a cancer treatment. It’s also a symbol of innovative biotechnology, and it has become a springboard for new therapies in autoimmune disease and infectious disease, too.
A CAR is a recombinant receptor that can reprogram T-cell function to target a specific antigen in cancer cells. It is composed of four parts, each of which contributes to the proper activation, functionality, and persistence of CAR-T cells.
① the extracellular antigen recognition domain of the single-chain fragment variant (scFv) derived from an antibody
② the hinge region (or spacer domain)
③ the transmembrane domain
④ the intracellular domain
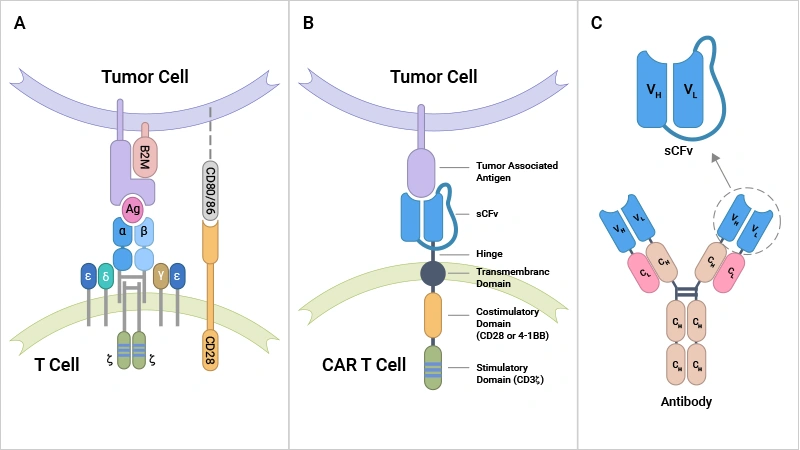
Figure 4. The differences in tumor antigen recognition between conventional TCRs and synthetic CARs [10]
Conventionally, T cell activation usually involves two signals: the first signal is initiated when the T-cell receptor (TCR) interacts with peptide-loaded major histocompatibility complex (pMHC), and the second signal comes from costimulatory receptors like CD28. The less-than-optimal effectiveness of conventional T cells in killing cancer cells may be partly caused by the absence of CD80/86 expression on tumor cells.
Differently, synthetic CARs can bind to antigens directly tumor-associated antigens (TAAs) on the surface of cancer cells through the scFv domain rather than relying on immunogenic processing and peptide presentation by MHC receptors, thus enhancing the applicability of CAR-T to different patients [11,12].
CARs have evolved. Currently, there are five generations of CAR-T cells.
Table 2. Five generations of CARs [13]
| Generations of CARs | Cytoplasmic Domain | Key Features | Clinical Stage |
|---|---|---|---|
| First-generation | Equipped with an extracellular antigen-recognizing domain combined with intracellular CD3ζ that mediates T cell activation | Provides T cell activation but not enough to sustain the therapeutic response. | not under investigation. |
| Second-generation | Equipped with an extracellular antigen-recognizing domain combined with two intracellular domains: CD3ζ and an additional costimulatory domain (e.g., CD28/4-1BB/ICOS/OX-40). | Co-stimulatory signal leads to higher activation, proliferation, cytokine secretion, and survival. | approved in some blood cancers, under investigation for solid tumors. |
| Third-generation | Equipped with an extracellular antigen-recognizing domain combined with three intracellular domains: CD3ζ and two additional costimulatory domains (e.g., CD28-4-1BB). | Multiple stimulatory signals enhance the activity profile without compromising safety—inconclusive long-term effects. | various phase 1/2 studies in blood cancers. |
| Fourth-generation | CD28 and 4-1BB co-stimulatory domains, NFAT inducible immune modulators, and CD3ζ (A diversified group of CAR-T constructs embracing armored CAR-T cells, cytokine-expressing CAR-T cells, switchable CAR-T cells, and universal CAR-T cells.). | Accompanying traditional CAR-T cell activity, the inducible secreted cytokines enhance proliferation and
persistence, leading to better anti-tumor capacity possible systemic cytotoxicity due to cytokine secretion in healthy tissues. |
phase 1/2 clinical studies. |
| Fifth-generation | CD28 and 4-1BB co-stimulatory domain, additional IL-2Rβ cytoplasmic domains, with STAT recruitment
motif and CD3ζ additional development in logic gated CARs. |
Has a higher stimulation capacity for T cells by activating the JAK-STAT pathway. | pre-clinical. |
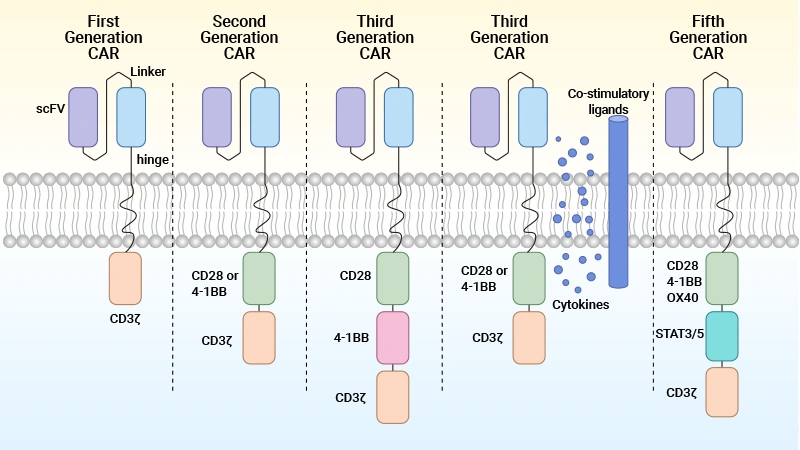
Figure 5. The structure and generation of the CAR-T cells [2]
How do CAR T-cells kill cancer? CAR-T cells mediate tumor killing through three axes:
① Perforin/Granzyme axis: When CAR-T cells recognize and bind to target tumor cells on the surface of cancer cells, they release perforin, which forms pores in the tumor cell membrane in a calcium-dependent manner. These pores allow granzymes to enter the tumor cell cytosol. Once inside, granzymes cleave key intracellular proteins, triggering apoptosis in the tumor cell. This pathway is particularly effective in targeting antigen-positive tumor cells.
② Fas and FasL axis (Death Receptor Pathway): CAR-T cells express FasL, which binds to Fas receptors on the surface of tumor cells. The Fas/FasL interaction activates the extrinsic apoptosis pathway, leading to the formation of the death-inducing signaling complex (DISC) and subsequent activation of caspases, which execute the apoptotic program in the tumor cell. This mechanism is particularly useful in cases where tumor cells may be resistant to other forms of cell death.
③ Cytokine secretion: Upon activation, CAR-T cells release cytokines such as IFN-γ, TNF-α, and IL-2, which directly induce apoptosis in tumor cells, enhance the cytotoxic activity of other immune cells, and promote an inflammatory microenvironment that is hostile to tumor growth. Additionally, cytokine production can recruit and activate other immune cells, amplifying the anti-tumor response.
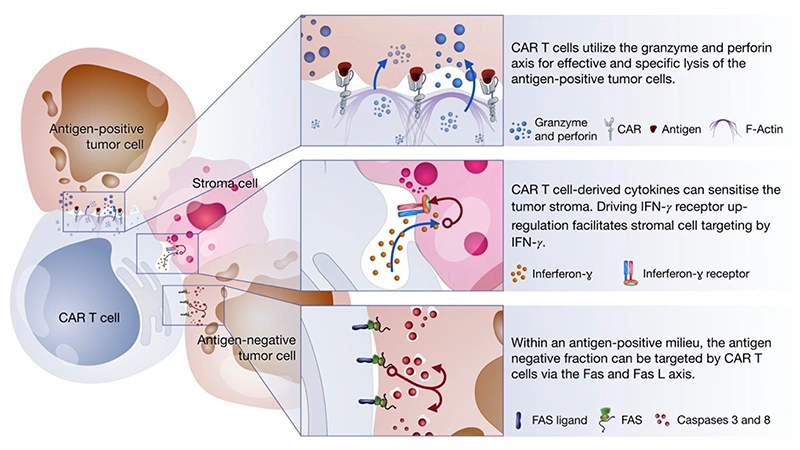
Figure 6. CAR-T cells-mediated tumor killing mechanisms [14]
The success of CAR-T therapy largely depends on identifying specific antigens expressed on cancer cells, which serve as "hot targets" for engineered T-cells to attack.
Hematological tumor:
Solid tumor:
Although CAR-T cell therapy represents a transformative approach to cancer treatment, several challenges and limitations hinder its broader application and efficacy.
Antigen escape occurs when tumor cells downregulate or lose the target antigen, rendering the therapy ineffective. This phenomenon is a significant barrier, as it allows cancer cells to evade immune detection and continue proliferating. Strategies to address this include targeting multiple antigens or developing CAR-T cells with broader specificity.
Many targets are not only on tumor cells. Even tiny amounts on normal tissue can be a problem. On-target off-tumor effects arise when CAR-T cells attack healthy tissues expressing the same antigen as the tumor. This can lead to severe organ damage and other adverse effects. For example, targeting CD19 in B-cell malignancies can also affect normal B cells, causing B-cell aplasia.
Antigen selection is closely correlated with sufficient anti-tumor efficacy and minimized toxicity concerns.
Effective CAR-T cell therapy requires the engineered cells to migrate to and infiltrate the tumor site. However, solid tumors often present physical and biological barriers, such as dense extracellular matrix and abnormal vasculature, which impede CAR-T cell trafficking. Improving CAR-T cell homing and penetration into tumors remains a critical area of research.
The tumor microenvironment (TME) is often highly immunosuppressive, with factors like regulatory T cells, myeloid-derived suppressor cells, and inhibitory cytokines that dampen CAR-T cell activity. Overcoming this immunosuppression is essential for enhancing CAR-T cell efficacy. Approaches include combining CAR-T therapy with immune checkpoint inhibitors or modifying CAR-T cells to resist suppression.
CAR-T cell therapy is associated with significant toxicities, mainly including cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). CRS, characterized by excessive cytokine release, can lead to life-threatening complications. ICANS is characterized by elevated cerebrospinal fluid cytokine levels and blood-brain barrier disruption and can manifest as confusion, seizures, or even coma. Managing these toxicities requires careful monitoring and the development of strategies to mitigate their impact.
The trick of balancing efficacy and toxicity in CAR-T cell therapy is to find the “sweet spot”—potent tumor killing, minimal collateral damage. This balancing act is the heart of every clinical trial and makes antigen selection, dosing, and patient monitoring critical.
Making CAR-T cells is complex. It takes time, precision, and quality control at every step.
Anti-CAR linker antibodies are used in research and development to monitor and detect CAR expression. Common linkers (like G4S and Whitlow) link the VL and VH chain of the scFv domain in the CAR. Special antibodies like anti-G4S linker mAb and anti-Whitlow linker mAb are used to detect these linkers and ensure correct CAR expression.
CUSABIO can offer anti-linker monoclonal antibodies to streamline preclinical and manufacturing QC processes.
| Product Name | Tested Applications |
|---|---|
| G4S linker Monoclonal Antibody (CSB-MA186062I1m) | ELISA, FC |
| G4S linker Monoclonal Antibody (CSB-MA186062I2m) | ELISA, FC |
| G4S linker Monoclonal Antibody (CSB-MA186062I3m) | ELISA, FC |
| G4S linker Monoclonal Antibody, FITC conjugated (CSB-MA186062I3m-C) | FC |
| Whitlow linker Monoclonal Antibody (CSB-MA234468A0m) | ELISA, WB, FC |
| Whitlow linker Monoclonal Antibodyy, FITC conjugated (CSB-MA234468A0m-C) | FC |
The field of immune cell therapies is rapidly evolving, with several innovative approaches showing promise in treating a range of diseases, including cancer and autoimmune disorders.
Engineered immune cell therapies are produced by initially collecting blood from the patient through apheresis, isolating T cells, and employing either viral or non-viral methods to introduce a transgene that encodes a synthetic receptor. Examples of engineered T cells include:
①T cells expressing an engineered TCR consisting of TCR alpha and beta subunits
② CAR-expressing T cells (CAR-Ts) or NK cells (CAR-NKs), which consist of an extracellular antigen-binding domain fused to intracellular domains involved in TCR signaling
③ CAAR T cells (CAAR-Ts), where the chimeric receptor is comprised of an antigen-binding domain that targets autoreactive B cells
④ CAR-Tregs (Regulatory T Cells), where Tregs are isolated from peripheral blood and engineered to express a CAR that redirects them to tissue affected by autoimmune disease
These cells keep the immune system in check. CAR-Tregs might help prevent rejection in organ transplants or treat autoimmune diseases by calming the immune system only where needed, reducing side effects common with regular immunosuppressants.
NK cells don't need to be "matched" to the patient, so therapy could be faster and safer. They act quickly, kill broadly, and are less likely to cause graft-versus-host disease or severe side effects. They can also be produced in large numbers, making commercial therapy easier.
Avoiding T-cell exhaustion is crucial for sustaining CAR-T cell efficacy.
T-cell exhaustion is driven by:
① Self-aggregation or excess antigen leading to constant stimulation.
② Tonic signaling via CD28, triggering exhaustion pathways.
③ Epigenetic modifications (via EZH2) reinforces the exhausted state.
④ Expression of exhaustion-related genes such as PD-1, TIM-3, LAG3, etc.
⑤ Activation of inhibitory receptors (e.g., PD-1, TIM-3) that suppress T-cell functions.
Potential solutions:
① Use of 4-1BB co-stimulation promotes expression of memory-related genes (LEF1, TCF7, IL7R, KLF2) and oxidative metabolism, supporting long-term T-cell function.
② Suppressing or modifying CD28 signaling to reduce tonic signaling.
③ Targeting EZH2 to prevent exhaustion-promoting epigenetic modifications.
④ Blocking inhibitory receptors (e.g., PD-1/PD-L1 inhibitors).
CAR-T cells engage with tumor cells expressing target antigens, leading to the release of soluble factors that trigger pro-inflammatory responses. This cascade activates myeloid cells, which subsequently produce key inflammatory cytokines including IL-6 and IL-1β. This eventually causes inflammatory toxicities.
Current management strategies for reducing inflammatory toxicities associated with CAR-T cell therapy include:
① Corticosteroids for broad immunosuppression;
② Tocilizumab (anti-IL-6 receptor antagonist) - currently the only FDA-approved therapy for CAR-T-associated CRS;
③ Anakinra (IL-1 receptor antagonist) shows promise in combination therapy.
The prevention strategies include:
① Genetic alterations to modify CAR-T cell function
② Multiple CAR targets to improve specificity
③ Enhanced CAR design to reduce off-target effects
④ Sequestration strategies (e.g., anti-GM-CSF)
⑤ Anti-inflammatory drugs (e.g., BTKIs)
⑥ Receptor antagonists (e.g., IL-1RA)
⑦ Transcriptional inhibition (e.g., JTE-607).
CAR-T cell therapy stands as a beacon of hope in the fight against cancer, embodying the transformative potential of personalized medicine. Its ability to reprogram the immune system to combat malignancies has not only saved lives but also redefined the boundaries of what is possible in oncology. However, as we celebrate its successes, the journey is far from over. Challenges such as high costs, manufacturing complexities, and managing severe side effects must be addressed to ensure broader accessibility and efficacy.
As research continues to push the frontiers of this innovative therapy, CAR-T cell treatment holds the promise of a future where cancer is no longer a death sentence but a manageable condition. The story of CAR-T is not just about scientific triumph—it’s about the enduring hope for a healthier, cancer-free world.
Further Reading:
T Cell Activation - The Switch for T Cell Executive Function
How the Tumor Microenvironment Fuels Cancer's Deadly Evolution
References
[1] Rosa, A., Upadhaya, S., Partridge, T., Shah, M., Correa, D., & Campbell, J. Landscape of cancer cell therapies: Trends and real-world data [J]. Nature Reviews Drug Discovery 21, 631-632 (2022).
[2] Luo, J., & Zhang, X. (2024). Challenges and innovations in CAR-T cell therapy: A comprehensive analysis [J]. Frontiers in Oncology, 14, 1399544.
[3] Kuwana Y, Asakura Y, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived c regions [J]. Biochem Biophys Res Commun (1987) 149:960–8.
[4] Eshhar, Z., Waks, T., et al. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors [J]. Proc. Natl. Acad. Sci. USA. 1993; 90:720-724.
[5] Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor [J]. Nat Biotechnol (2002) 20:70–5.
[6] Imai C, Mihara K, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia [J]. Leukemia (2004) 18:676–84.
[7] Kershaw MH, Westwood JA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer [J]. Clin Cancer Res (2006) 12:6106–15.
[8] Lamers CH, Sleijfer S, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience [J]. J Clin Oncol (2006) 24:e20–22.
[9] Munshi NC, Anderson LD, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma [J]. N Engl J Med (2021) 384:705–16.
[10] Mitra, A., Barua, A., et al. (2023). From bench to bedside: The history and progress of CAR T cell therapy [J]. Frontiers in Immunology, 14, 1188049.
[11] Zhao ZG, Condomines M, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells [J]. Cancer Cell. (2015) 28:415–28.
[12] Sadelain M, RiviSre I, Riddell S. Therapeutic T cell engineering [J]. Nature. (2017) 545:423–31.
[13] Patel, Kisha K. et al. (2025) From concept to cure: The evolution of CAR-T cell therapy [J]. Molecular Therapy, Volume 33, Issue 5, 2123 - 2140.
[14] Benmebarek, M., Karches, et al. (2018). Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells [J]. International Journal of Molecular Sciences, 20(6), 1283.


Comments
Leave a Comment