Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
On March 17, 2022, TODD R. GOLUB's team from Harvard University and MIT published a research paper on the Science entitled "Copper induces cell death by targeting lipoylated TCA cycle proteins". The scientists discover a new kind of cell death linked to copper--naming it "Cuprotosis".
Cell death research has made great improvements over the past decades. It is not just unraveling the mechanisms of fundamental biological processes, but also leading to new therapies for cancer and other diseases such as certain autoimmune pathologies or infectious diseases. Among them, Apoptosis, Pyrolysis, Necrosis and Ferroptosis are the most extensively studied forms of cell death in recent years (Click to read "Cell Biology" feature articles). Here, we will learn about the latest reports of copper-induced cell death--"Cuprotosis".
The TODD R research team revealed that "Cuprotosis" is a novel form of cell death--copper-dependent cell death. Copper directly bind to the thioctylated components of the tricarboxylic acid (TCA) cycle, leading to abnormal aggregation of thioctylated proteins and decreased expression of Fe-S cluster proteins, resulting in a proteotoxic stress response and a distinct form of cell death.
Speaking of which, it means that a more effective copper carrier therapy can be developed for the certain molecule, especially for FDX1, tumor types with high levels of lipoic acidification and reliance on mitochondrial respiration. Thus, "Cuprotosis" as the new kind of cell death, caused a great deal of excitement among scientists!
In the study, TODD R team provides insight into the mechanism of a copper-triggered cell death. To learn how the team discovered this new form cell death "Cuprotosis" and how its potential mechanisms, we bring the six results analysis process as follows.
Firstly, TODD R team found that all copper ionophores can kill cells. And the use of the copper ionophores Elesclomol can regulate copper-mediated cell death. But, is this cell death distinct from the traditional modes of cell death (like apoptosis, necrosis, and ferroptosis)? To further verify this, knock down experiments were performed on the key effectors of apoptosis by Bax and BAK1, both of which failed to prevent copper-induced cell death. Apparently, it indicates that the cell death mechanism is different from the well-recognized of cell death pathway. In a nutshell, these data confirm that copper ionophores induce a distinct form of regulated cell death.
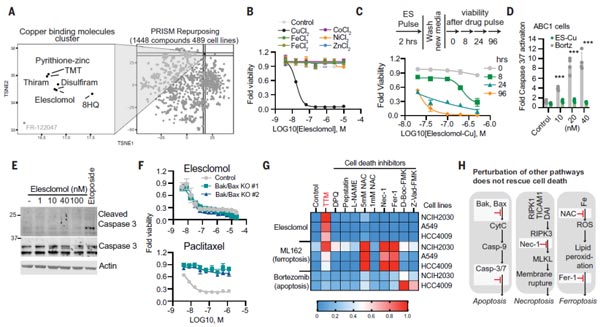
Figure 1. Copper ionophores induce a distinct form of regulated cell death [1].
Immediately, the TODD R team found that mitochondrial respiration plays a key role in this death mechanism. The experimental data showed that when cells are dependent on mitochondrial respiration, their sensitivity to copper ionophores is nearly 1,000-fold higher than that of cells undergoing glycolysis; treatment of cells with mitochondrial antioxidants, fatty acids, and inhibitors of mitochondrial function significantly affected the sensitivity of cells to copper ionophores.
Besides, the team observed that the mitochondrial depolymer FCCP had no effect on copper ionophores-induced cell death; the team then examined the effects of different types of hypoxic stimuli to cells, versus copper ionophores treatment on cellular oxygen consumption rate (OCR), which suggest that copper does not directly target the electron transport chain (ETC), but exerts effects only in the tricarboxylic acid (TCA) cycle. These observations suggest that it is mitochondrial respiration that is necessary for copper ionophores to induce cell death.
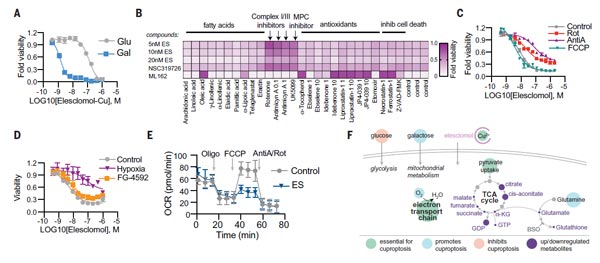
Figure 2. Mitochondrial respiration regulates copper ionophores-induced cell death [1].
To further clarify the metabolic pathways of cell death, the team used a genome-wide CRISPR-Cas9 loss-of-function screens with differently structured copper ionophores to improve the generalizability of the universal screening. The team performed independent knockdown experiments to select of 3,000 metabolic enzymes.
From the above screening process, they found seven positively regulated genes (FDX1, LIAS, LIPT1, DLD, DLAT, PDHA1, and PDHB), and three negatively regulated genes (MTF1, GLS and CDKN2A), of which the ferredoxin 1 gene (FDX1) and protein lipoylation are key factors of copper ionophores-induced cell death. Beyond this, deletion of FDX1 and lipoic acid synthase (LIAS) genes enhanced resistance to copper-induced cell death via knockdown experiments, which implied a strong link between FDX1 and protein lipoylation and copper ionophores-induced cell death. These studies suggest that FDX1 and protein lipoylation are key mediators of copper-induced cell death.
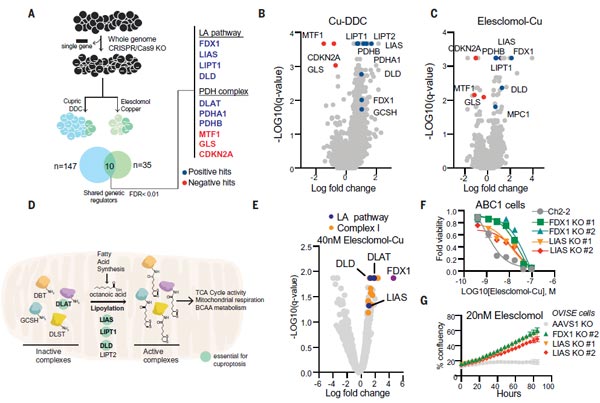
Figure 3. FDX1 and protein lipoylation are key mediators of copper ionophores-induced cell death [1].
Subsequently, the team found that knockdown of either FDX1 or the protein lipoylation-related enzymes could protect cells from copper toxicity. Thus they speculated whether FDX1 could be an upstream regulator of protein lipoic acidification. Using public databases such as the Cancer Dependency Map, they found that FDX1 and protein lipoylation-related enzymes are highly correlated with cell survival.
Afterwards, they performed immunohistochemical staining analysis of FDX1 and lipoic acid pathway proteins in 208 human tumor specimens, which showed that the expression of FDX1 and lipoic acidylated proteins were highly correlated; WB experiments further confirmed that knockdown of FDX1 resulted in the loss of protein lipoylation of DLAT (Dihydrolipoamide S-Acetyltransferase) and DLST (Dihydrolipoamide S-Succinyltransferase); metabolite analysis revealed that FDX1 deletion leads to the accumulation of pyruvate and α-ketoglutarate, succinate depletion, and accumulation of S-adenosylmethionine (SAM), a key substrate of LIAS. The above data confirm that FDX1 is involved in the protein lipoylation.
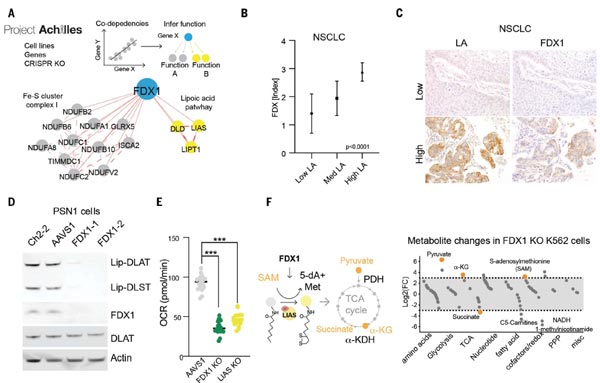
Figure 4. FDX1 is an upstream regulator of protein lipoylation [1].
Based on the previous experiments, the scientists established a link between copper toxicity and protein lipoylation. Actually, it only indirectly identified the importance of them in copper induced-cell death, but without establishing a direct mechanistic link. Does copper bind directly to lipidated proteins?
To test this hypothesis, they performed a series of analyses. First, the team purified two proteins, DLAT and DLST in lysates from FDX1 knockout cells and normal cells. It indicted that DLAT and DLST in normal cells bind directly to copper (Cu), but not to cobalt (Co) or nickel (Ni) resins. In contrast, DLAT and DLST in FDX1 knockdown cells do not bind copper, indicating that the lipoyl moiety is required for copper binding.
Furthermore, treatment of Elesclomol-sensitive cells increased the level of DLAT oligomers, whereas treatment of Elesclomol-insensitive cell lines or FDX1 knockdown resulted in DLAT oligomerization only at high concentrations of Elesclomol. It was further revealed by mass spectrometry analysis that copper ionophores treatment also resulted in reduced levels of Fe-S cluster proteins. The above data suggest that copper directly binds and induces the oligomerization of lipoylated DLAT.
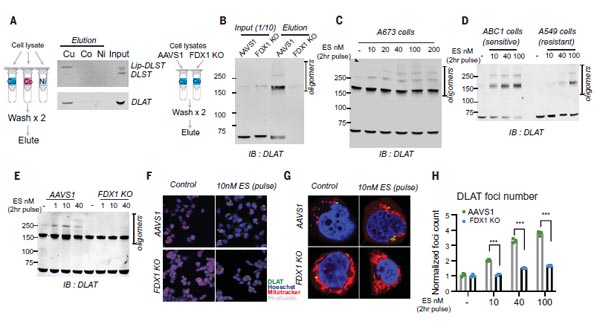
Figure 5. Copper directly binds and induces the oligomerization of lipoylated DLAT [1].
At this point, the mechanism of copper death has been uncovered through the validation of above 1-5: copper-dependent death occurs by means of direct binding of copper to lipoylated components of the tricarboxylic acid (TCA) cycle. This results in lipoylated protein aggregation and subsequent iron-sulfur cluster protein loss, which leads to proteotoxic stress and ultimately cell death. However, it is unclear whether the mechanisms of copper toxicity associated with copper ionophore treatment are shared by these naturally occurring disorders of copper homeostasis.
To explore the potential connection, the authors first overexpressed the copper transporter SLC31A1 (a key target of copper homeostasis) in human embryonic kidney HEK 293T and ABC1 cells, suggesting that copper input significantly enhanced total cellular depletion of mitochondrial respiration-related proteins, reduced protein lipoylation, reduced levels of Fe-S cluster proteins, and increased HSP70 stress protein levels.
Of note, ferroptosis, necroptosis, and apoptosis inhibitors did not prevent copper-induced cell death in cells overexpressing SLC31A1, but copper chelator, FDX1 knockdown and LIAS knockdown attenuated the cell death phenomenon.
At last, Wilson's disease mouse model was applied, in which Atp7b-deficient mice leads to the loss of lipoylated and Fe-S cluster proteins, as well as an increase in HSP70 abundance. These findings in mouse models of copper toxicity suggest that copper overload results in the same cellular effects as those induced by copper ionophores.
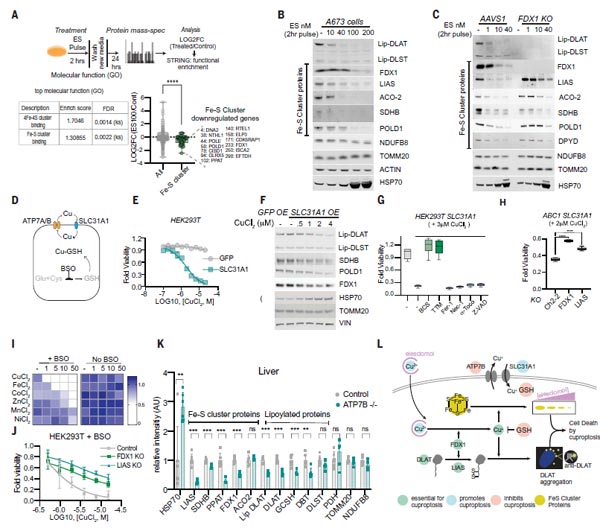
Figure 6. Copper overload results in the same cellular effects as those induced by copper ionophores [1].
At the present study, the authors discover a new kind of cell death linked to copper--defined as "Cuprotosis". Notably, they determined a series of key genes mainly responsible for the "Cuprotosis" regulation mechanism, which will be a hot topic for future research in diseases caused by disorders of copper, including Wilson’s disease, Menke’s disease, and tumors. CUSABIO has summarized the hot targets of antibody products in the "Cuprotosis" regulation mechanism to support your research on "Cuprotosis" (See table below).
A Summary of Hot Targets of Antibody Products in the "Cuprotosis"
| Pathways | Regulation method | Key Targets | Code | Hosts | Reaction Species | Applications |
|---|---|---|---|---|---|---|
| Lipoic acid pathway | Positive regulation | DLD | CSB-PA006928LA01HU | Rabbit | Human, Mouse, Rat | ELISA, WB, IHC |
| CSB-PA783949 | Rabbit | Human, Mouse, Rat | ELISA, IHC | |||
| DLAT | CSB-PA006926LA01HU | Rabbit | Human | ELISA, WB, IP | ||
| CSB-PA445587 | Rabbit | Human, Mouse, Rat | ELISA, WB, IHC | |||
| Pyruvate dehydrogenase (PDH) complex pathway | LIAS | CSB-PA012927LA01HU | Rabbit | Human | ELISA, WB, IHC | |
| PDHA1 | CSB-PA017715LA01HU | Rabbit | Human | ELISA, IHC, IF | ||
| CSB-PA247984 | Rabbit | Human | ELISA, WB, IHC | |||
| PDHB | CSB-PA017717ESR2HU | Rabbit | Human | ELISA, IHC | ||
| CSB-PA017717GA01HU | Human, Mouse, Rat | Rabbit | ELISA, WB, IHC | |||
| Negative regulation | GLS (GAM/GAC/KGA) | CSB-PA009528DA01HU | Human, Mouse | Rabbit | ELISA, WB, IHC | |
| CSB-PA969762 | Human, Mouse, Rat | Rabbit | ELISA, WB, IHC | |||
| CSB-PA177709 | Human, Mouse, Rat | Rabbit | ELISA, IHC | |||
| MTF1 | CSB-PA013754LA01HU | Human | Rabbit | ELISA, WB, IHC, IF | ||
| P16-INK4A (CDKN2A) | CSB-PA005089LA01HU | Human | Rabbit | ELISA, IHC | ||
| CSB-PA003618 | Human, Mouse | Rabbit | WB, ELISA | |||
| Glycine Cleavage System (GCS) system pathway | / | GCSH | CSB-PA009335GA01HU | Human, Mouse, Rat | Rabbit | ELISA, WB, IHC, IF |
Reference
1. Tsvetkov, Peter, et al. "Copper induces cell death by targeting lipoylated TCA cycle proteins." science 375.6586 (2022): 1254-1261.
Popular Topics
Related Articles