Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
In 1945, when K. R. Porter and A. Claude et al. observed cultured mouse fibroblasts, they found a network structure in the cytoplasm, which was later named endoplasmic reticulum (ER).
The endoplasmic reticulum is a continuous membranous system that forms a network of interconnected vesicles surrounded by flat membranous sacs or tubular structures. The endoplasmic reticulum exists in most eukaryotic cells except for mature red blood cells in mammals.
There are two types of endoplasmic reticulum: rough endoplasmic reticulum (RER) and smooth endoplasmic reticulum (SER).
The rough endoplasmic reticulum is a flattened sac, neatly arranged with ribosomes embedded on its surface, which is the site of protein synthesis.
The smooth endoplasmic reticulum is branched tubular or vesicular without ribosome attachment. The smooth endoplasmic reticulum synthesizes almost all lipids, including phospholipids and cholesterol. Smooth endoplasmic reticulum also has the function of detoxification and glycogen metabolism. Cells that specialize in producing proteins have more rough endoplasmic reticulum, while cells that produce lipids (fats) and steroid hormones have more smooth endoplasmic reticulum. Cells do not contain pure rough or smooth endoplasmic reticulum; they are part of a continuous structure of the endoplasmic reticulum, respectively.
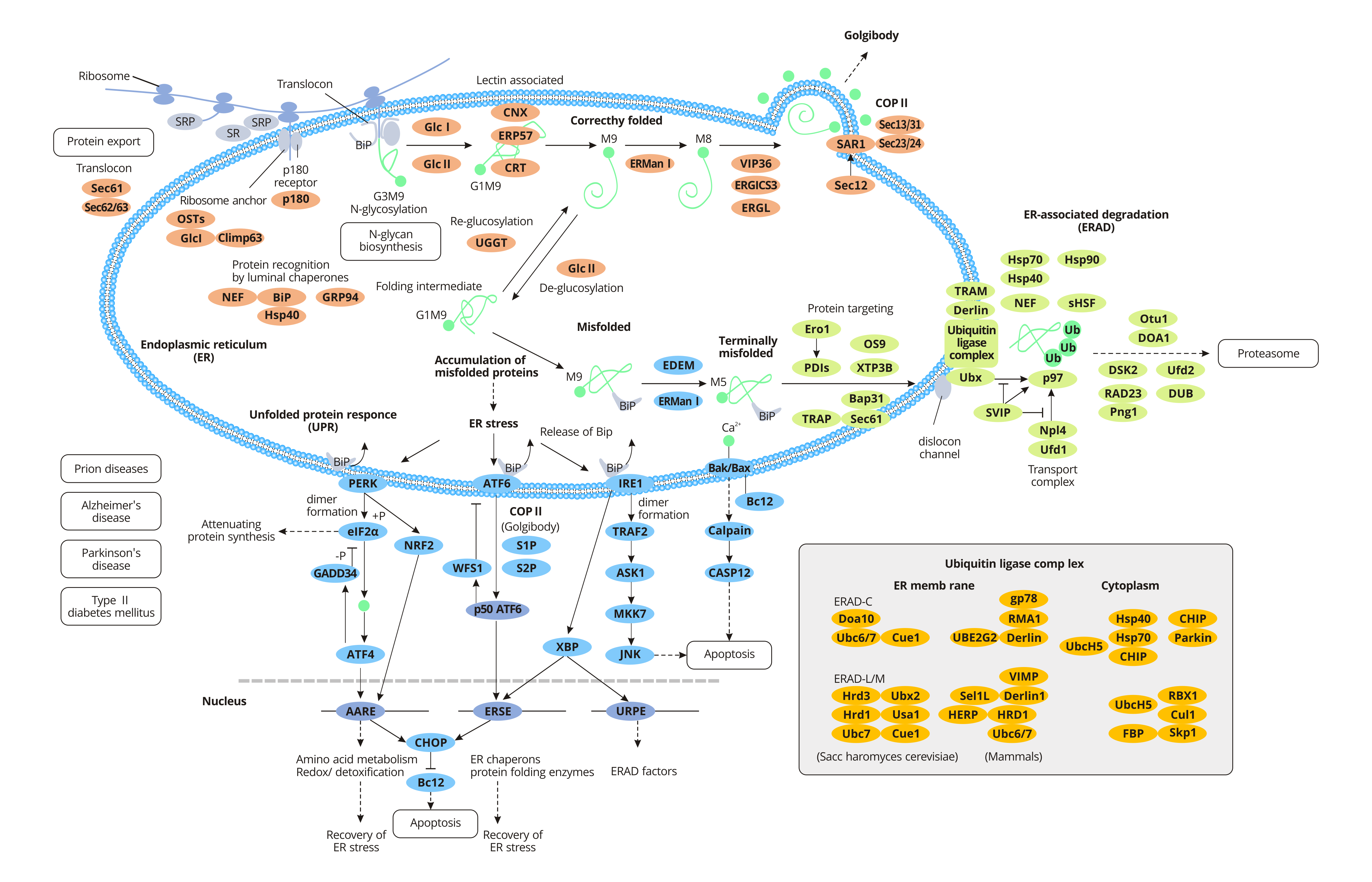
For specific secreted proteins, polypeptide chains need to be modified in different ways post-translation to obtain physiological functions. The newly synthesized peptides are transported to the endoplasmic reticulum and folded into the correct three-dimensional structure before being transported to their destination. When proteins are improperly processed or modified, the endoplasmic reticulum acts as a quality control agent, trapping the non-folded or misfolded proteins in the endoplasmic reticulum and clearing them through the ubiquitin-proteosome pathway (UPP).
Some proteins are produced on free ribosomes in the cytoplasm, which remain in the cytoplasmic matrix or are transported to the nucleus, mitochondria, and peroxidases. Other proteins, such as secretory proteins, integral membrane proteins, and soluble resident proteins in organelles, are transferred to the endoplasmic reticulum along with ribosomes shortly after the initial synthesis. Nascent peptides pass through the endoplasmic reticulum and enter the endoplasmic reticulum lumen in the rough endoplasmic reticulum.
The translation of secretory or integrin proteins initiates in the cytoplasm, and the ribosomes containing these mRNAs are identified by signal recognition particles (SRPs) through a signal sequence called signal peptides within the amino terminus of the newly synthesized peptides. Once SRP binds to ribosomes with new peptides, the extension of the peptide chain is terminated. After that, the SRP-ribosome complex moves to the endoplasmic reticulum and binds to the SRP receptor on the endoplasmic reticulum, restarting the extension of the peptide chain. At the same time, SRP is released into the cytoplasmic matrix and rebinds to other signal peptides. The signal peptide guides the new peptide to enter the rough endoplasmic reticulum lumen through transposon co-translation. The transposon is a channel containing several Sec proteins (Sec61, Sec62/Sec63) across the lipid double membrane. After the signal peptide enters the endoplasmic reticulum, it is cut off from the peptide chain by signal peptidase and quickly degraded by other proteases.
Proteins synthesized in the rough endoplasmic reticulum fold correctly with the help of endoplasmic reticulum lumen chaperone BiP and are packaged into transport vesicles for transport to the Golgi apparatus. Bip, a member of the heat shock protein 70 family (HSP70), promotes proper protein folding. During protein folding, glycosylation occurs with the action of related proteins, in which oligosaccharide chains are attached to the asparaginic acid of the peptide chain. Most proteins synthesized by endoplasmic reticulum are glycosylated. Glycosylation helps some proteins fold correctly, resists the action of digestive enzymes and confers them the ability to conduct signals. The formation of disulfide bonds in protein molecules is closely related to the folding of new peptides and is very important to maintain the structural stability and function of protein molecules.
Protein disulfide bond isomerase (PDI) is abundant in the endoplasmic reticulum and catalyzes the exchange reaction between the sulfhydryl group and disulfide bond in protein molecules. The formation of disulfide bonds strengthens the spatial structure of proteins and further stabilizes their conformation. And the formation of disulfide bonds is necessary for the activity of many enzymes and proteins. Misfolded proteins are retained in the endoplasmic reticulum lumen to form complexes with molecular chaperones. In general, misfolded proteins at the end bind to BiP and then are degraded by the proteasome, a process known as endoplasmic reticulum-associated degradation (ERAD). Accumulation of misfolded proteins in the endoplasmic reticulum leads to endoplasmic reticulum stress (ERS) and activates a signaling pathway called the unfolded protein response (UPR). However, in some severe cases, the protective mechanism activated by UPR is insufficient to restore normal ER function and ultimately triggers apoptosis and death.
Misfolded proteins that are neither protected by molecular chaperones nor degraded by proteases may interact with each other to form aggregates. These aggregates, which result from the failure of endoplasmic reticulum quality control, disrupt the balance of cells and aggregate other unwanted proteins, thus causing some diseases.
Bovine spongiform encephalopathy (BSE) is a group of neuropathic lesions caused by prion protein (PrP) in humans and animals. Natural PrP is rich in alpha-helix, namely PrPc, and it is not pathogenic. An unknown protein converts the PrPc into a beta-folded PrPsc, causing disease.
When alpha 1 antitrypsin (SERPINA1) is mutated, only 15 percent of the protein is secreted, and the rest remains in the endoplasmic reticulum. This retention is due in part to calnexin (CANX), a molecular chaperone in the endoplasmic reticulum, which mediates the aggregation of mutated proteins that misfold. The accumulation of abnormal products greatly hinders the normal activities of cells, leading to the occurrence of cirrhosis or emphysema. Alzheimer's disease (AD) is a disorder in which the brain is filled with clumps of misfolded proteins called amyloid-beta plaques and tau proteins.

