Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
CD4⁺ T cells are central players in the adaptive immune system, orchestrating immune responses against diverse pathogens and maintaining immune homeostasis. Based on their differentiation pathways and functional properties, CD4⁺ T cells were historically classified into T helper 1 (Th1), Th2, and regulatory T (Treg) cells. However, the discovery of Th17 cells—a distinct lineage specialized in producing IL-17—revolutionized our understanding of CD4⁺ T cell biology. Th17 cells differentiate from naive CD4⁺ T cells under specific cytokine conditions and play critical roles in autoimmune diseases, infectious diseases, and cancer [1].
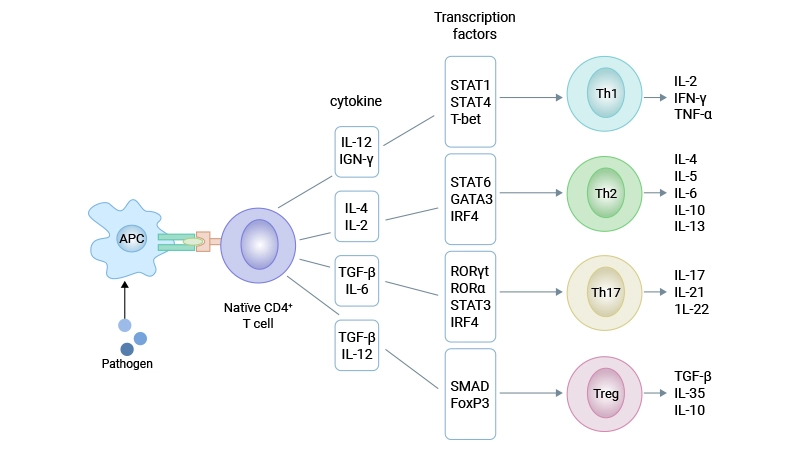
Figure 1. Four subsets of CD4⁺ T cells
This article walks through the discovery, markers, cytokine outputs, differentiation signals, transcriptional control, immunomodulation, disease links, and therapeutic targeting of Th17 cells, with an emphasis on concepts that bridge basic immunology and translational applications.
Table of Contents
3. Differentiation Regulation of Th17 Cells
4. Biological Effects of Th17 Cells
5. Dysregulation of Th17 Cell Differentiation-Related Diseases
For decades, the Th1/Th2 paradigm dominated our understanding of CD4⁺ T cell differentiation. This framework explained many immune responses but failed to account for certain autoimmune and inflammatory conditions. A breakthrough came in 2003 when researchers showed that IL-23, not IL-12, was essential for autoimmune inflammation in the brain [3].
This finding led to the formal identification of Th17 cells in 2005 by two independent research groups, which established Th17 as a distinct lineage from Th1 and Th2 cells [1,2]. Using mouse models of experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (CIA), they confirmed that Th17 cells are the primary drivers of tissue inflammation in these autoimmune diseases. The discovery of Th17 cells provided a new therapeutic target for previously poorly understood autoimmune disorders.
Accurate identification of Th17 cells is essential for studying their biology and clinical relevance. Th17 cells can be distinguished from other CD4⁺ T cell subsets using a combination of extracellular surface markers and intracellular transcription factors and cytokines.
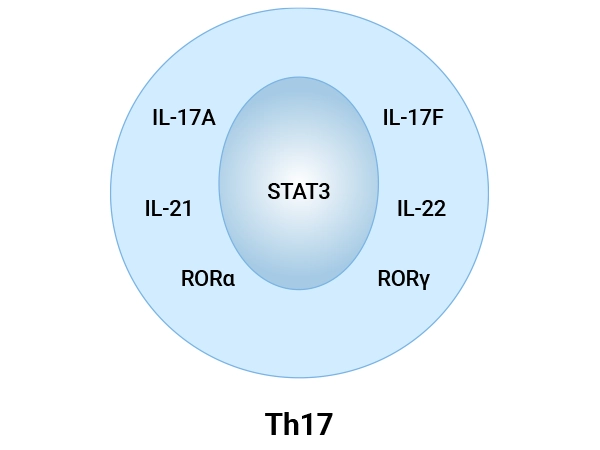
Figure 2. Cell Markers of Th17 Cells
In humans, the most reliable surface markers for identifying Th17 cells are:
Intracellular markers are more specific for Th17 cells and include both transcription factors and effector cytokines:
RORγt (RORC): The lineage-defining orphan nuclear receptor, encoded by the RORC gene, is essential for initiating Th17 cell differentiation and driving the expression of signature cytokines like IL-17 [23].
RORα: Plays a complementary and critical role, particularly in the differentiation of pathogenic Th17 (pTh17) cells. Studies show that RIP2 deficiency enhances pTh17 cell formation in a manner dependent on RORα.
STAT3: STAT3 is vital for transducing cytokine signals (e.g., from IL-6, IL-21, IL-23) during Th17 differentiation. Its activity is profoundly diminished in neonatal T cells, contributing to their impaired Th17 responses
IL-17A and IL-17F: These are the hallmark cytokines of the Th17 lineage, directly responsible for recruiting neutrophils, promoting inflammation, and contributing to tissue remodeling and autoimmune pathology.
IL-21: An autocrine cytokine that amplifies Th17 cell differentiation and stabilizes the lineage.
IL-22: Often co-produced with IL-17, IL-22 has immunoregulatory and tissue-protective functions. Notably, neonatal Th17 responses are skewed towards a high IL-22 phenotype, which may represent a developmental adaptation.
GM-CSF (CSF2): Produced by pathogenic Th17 subsets and strongly associated with their encephalitogenic potential in diseases like multiple sclerosis.
The differentiation of Th17 cells is a tightly regulated process controlled by a complex network of cytokines, signaling pathways, and transcription factors. Unlike Th1 and Th2 cells, which are driven by single master cytokines (IL-12 for Th1, IL-4 for Th2), Th17 cell differentiation requires the coordinated action of multiple cytokines.
Th17 cell differentiation proceeds through three sequential stages: induction, amplification, and stabilization. Distinct cytokines and signaling pathways regulate each stage.
The initial differentiation of naive CD4⁺ T cells into Th17 cells requires the synergistic action of TGF-β and IL-6 [5].
TGF-β A shared driver of both Th17 and Treg differentiation, with the outcome shaped by the presence or absence of pro‑inflammatory cytokines such as IL‑6 [6]. In low‑inflammation settings, TGF‑β favors FOXP3⁺ Treg induction, which can suppress immune responses, whereas in the presence of IL‑6 or IL‑1β, the balance shifts toward RORγt expression and Th17 differentiation [33,34].
Th17 and Treg lineages show reciprocal plasticity: under inflammatory conditions, some Tregs can downregulate FOXP3 and acquire IL‑17 expression, while Th17 cells can gain regulatory features, including IL‑10 production, in tolerogenic environments.
IL-6 The key cytokine that initiates Th17 lineage commitment. IL-6 signals through the gp130 receptor subunit to activate STAT3, which directly binds to the promoters of Rorc (encoding RORγt) and Il17 genes [22]. The IL-6-gp130-STAT3 pathway is absolutely required for Th17 cell differentiation.
IL-1 Plays a critical role in the early stages of Th17 differentiation. In the absence of exogenous TGF-β, IL-1 can synergize with IL-6 and IL-23 to promote Th17 cell development. IL-1 also enhances the expression of IL-23R on differentiating Th17 cells [8].
Once initiated, Th17 cell differentiation is amplified by IL-21, which is secreted by Th17 cells themselves in a STAT3-dependent manner [9].
IL-21 forms an autocrine positive feedback loop: it activates STAT3 in differentiating Th17 cells, which further induces IL-21 production and upregulates IL-23R expression. This loop dramatically increases the number of Th17 cells and enhances their effector function. In the absence of IL-6, IL-21 can even substitute for IL-6 to drive Th17 differentiation when combined with TGF-β [7].
The final stage of Th17 cell differentiation is mediated by IL-23. Although IL-23 is not required for the initial induction of Th17 cells, it is essential for their terminal differentiation, survival, and maintenance of pathogenic function [24].
IL-23 signals through its receptor (composed of IL-23R and IL-12Rβ1 subunits) to activate the JAK-STAT pathway, primarily STAT3. This signaling pathway stabilizes the Th17 phenotype and promotes the production of pro-inflammatory cytokines, including IL-17A, IL-17F, and IL-22. Without IL-23 signaling, Th17 cells fail to become pathogenic and cannot induce autoimmune disease [4].
Several transcription factors work together to drive Th17 cell differentiation:
STAT3 The central signaling molecule that integrates signals from multiple cytokines (IL-6, IL-21, IL-23) and directly activates the expression of RORγt, IL‑17A, IL‑17F, IL‑21, and IL‑23R, among other genes involved in proliferation and survival of Th17 cells [10,12,15,31].
Genome‑wide binding analyses show that STAT3 occupies regulatory regions of many Th17‑associated genes and coordinates with other factors such as IRF4, BATF, and c‑Maf to establish the Th17 transcriptional network [31,32].
RORγt The lineage-defining transcription factor for Th17 cells. RORγt directly activates IL17A and IL17F gene transcription and is required for the development and maintenance of Th17 cells in mice and humans [29,32].
Continuous expression of RORγt in mature Th17 cells helps safeguard their lineage specification and prevents conversion to alternative fates such as Th2 cells. This central role makes RORγt an attractive drug target for selectively dampening Th17 responses [29,30].
RORα Works synergistically with RORγt to enhance Th17 cell differentiation.
IRF4 and BATF: Pioneer‑like factors that open chromatin at Th17‑associated loci and cooperate with RORγt and STAT3 to enhance IL‑17 expression [11,22,31,32].
Runx1 and HIF‑1α: Promote Th17 differentiation and support metabolic programs that favor glycolysis in inflammatory Th17 cells [32].
FOXP3 The signature transcription factor of regulatory T cells (Tregs), which can antagonize RORγt and suppress the Th17 program when induced in the absence of strong inflammatory cues [33].
The relative activity of these factors, influenced by cytokines and environmental signals, determines whether a naïve CD4⁺ T cell becomes a Th17 cell, a Treg, or adopts another helper fate.
To prevent excessive inflammation and maintain immune homeostasis, Th17 cell differentiation is tightly controlled by multiple negative regulators. These factors counteract the pro-differentiation signals and limit the magnitude of Th17-mediated immune responses.
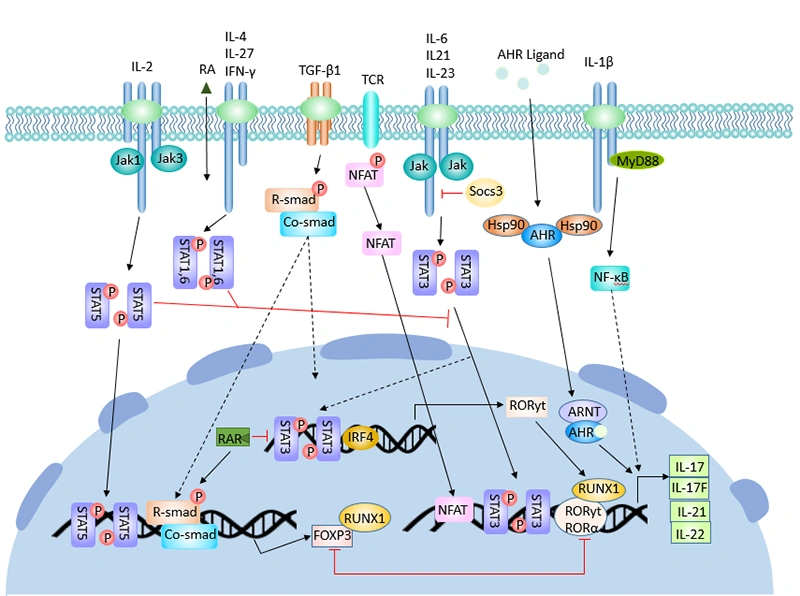
Figure 3. The regulatory process of Th17 cell differentiation
Th17 cells are sensitive to metabolic and environmental cues, including hypoxia, nutrient availability, and microbiota‑derived metabolites. HIF‑1α and related pathways promote glycolytic metabolism that supports inflammatory Th17 phenotypes, whereas signals that favor oxidative phosphorylation may support more regulatory or less pathogenic Th17 states [32,34,37].
Short‑chain fatty acids from commensal bacteria, vitamin A derivatives, and aryl hydrocarbon receptor ligands are among the metabolites that influence Th17/Treg balance in mucosal tissues [34,37].
Th17 cells exert their biological effects primarily through the secretion of a unique panel of pro-inflammatory cytokines. These cytokines act on various cell types to coordinate immune responses against extracellular pathogens and mediate tissue inflammation.
The most important effector cytokine produced by Th17 cells is IL-17, which belongs to a family of six structurally related cytokines (IL-17A to IL-17F) [17]. IL-17 signals through a receptor complex composed of IL-17RA and IL-17RC subunits, which are expressed on a wide range of cells, including epithelial cells, endothelial cells, fibroblasts, and immune cells [25].
The main biological functions of IL-17 include:
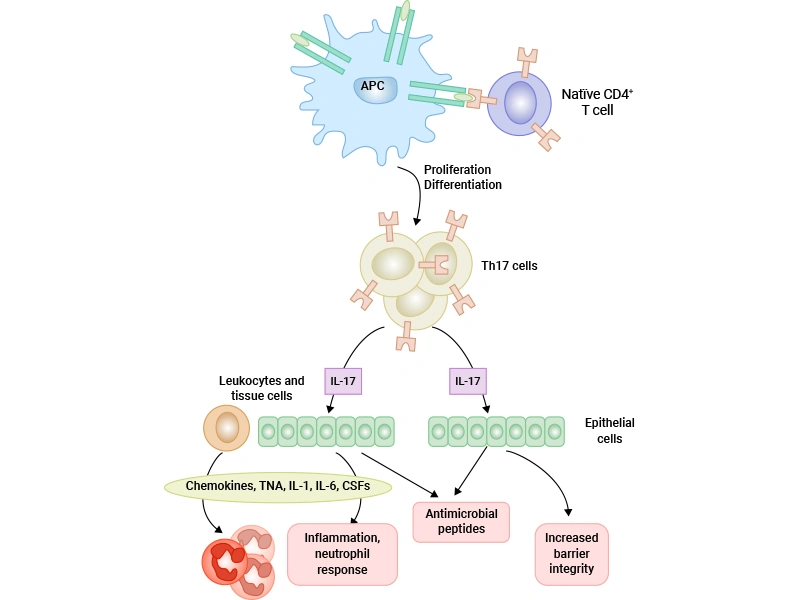
Figure 4. Biological Effects of Th17 cells
In addition to IL-17, Th17 cells also secrete IL-21 and IL-22. IL-21 regulates B cell differentiation and antibody production, while IL-22 acts on epithelial cells to enhance barrier function and promote tissue repair. Together, these cytokines create a powerful inflammatory environment that is essential for clearing extracellular bacterial and fungal infections.
Given their potent effector functions, it is not surprising that dysregulated Th17 responses are implicated in diverse diseases. Understanding these associations can help link mechanistic experiments to clinical phenotypes and guide the selection of suitable disease models.
Th17 cells and IL‑17 family cytokines are strongly associated with:
Th17 cells are essential for host defense against extracellular bacteria and fungi, particularly those that infect mucosal surfaces. They play a critical role in immunity against Staphylococcus aureus, Klebsiella pneumoniae, and Candida albicans [27].
During infection, dendritic cells produce IL-6, IL-1β, and IL-23, which drive Th17 cell differentiation. Th17 cells then migrate to the site of infection and secrete IL-17 and IL-22, which recruit neutrophils and enhance epithelial barrier function. Defects in Th17 cell development or function result in severe and recurrent mucocutaneous candidiasis and bacterial infections.
Emerging data link Th17 cells to cancer, with context‑dependent roles in tumor promotion or suppression. In some settings, Th17‑derived cytokines support tumor growth by promoting angiogenesis and chronic inflammation, whereas in others they enhance antitumor immunity through recruitment of effector cells.
Most evidence supports a pro-tumorigenic role for Th17 cells. IL-17 promotes tumor growth by inducing angiogenesis, suppressing anti-tumor immune responses, and enhancing tumor cell invasion and metastasis [19,20]. High numbers of tumor-infiltrating Th17 cells are associated with poor prognosis in many human cancers, including gastric, breast, and lung cancer.
However, some studies have shown that Th17 cells can also exert anti-tumor effects. In certain contexts, Th17 cells can recruit and activate cytotoxic T cells and natural killer (NK) cells, leading to tumor regression [21,28]. The dual role of Th17 cells in cancer highlights the need for further research to fully understand their functions in different tumor microenvironments.
The strong clinical and mechanistic evidence linking Th17 cells to disease has spurred the development of therapies targeting IL‑17, IL‑23, or upstream regulators. These agents also provide “experiments in humans” that refine our understanding of Th17 biology.
Several monoclonal antibodies targeting IL‑17A, IL‑17 receptor A, or IL‑23/IL‑12p40 are approved for psoriasis and related conditions:
Meta‑analyses indicate that both IL‑17 and IL‑23 inhibitors are highly effective and generally well tolerated in psoriasis, though IL‑17 blockade may be associated with a slightly higher rate of adverse events in some studies.
Beyond neutralizing cytokines, several strategies aim to indirectly modulate Th17 responses:
These approaches are at various stages of preclinical and clinical development and may offer more nuanced control of Th17‑driven pathology.
The discovery of Th17 cells has fundamentally transformed our understanding of CD4⁺ T cell biology and immune regulation. Th17 cells are a distinct lineage of helper T cells that differentiate under the control of TGF-β, IL-6, and IL-23, and are characterized by the expression of RORγt and the production of IL-17. They play essential roles in host defense against extracellular pathogens but are also major drivers of autoimmune inflammation when dysregulated.
Over the past two decades, significant progress has been made in elucidating the molecular mechanisms that regulate Th17 cell differentiation and function. This knowledge has led to the development of novel therapeutic strategies targeting the IL-23/Th17 pathway for the treatment of autoimmune diseases. However, many questions remain, particularly regarding the plasticity of Th17 cells and their complex roles in cancer and infectious diseases. Continued research in this field will undoubtedly lead to new insights and improved therapies for a wide range of human diseases.
References
[1] Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17 [J]. Nature Immunology. 2005;6(11):1133-1141.
[2] Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages [J]. Nature Immunology. 2005;6(11):1123-1132.
[3] Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain [J]. Nature. 2003;421(6924):744-748.
[4] Croxford AL, Mair F, Becher B. IL-23: One cytokine in control of autoimmunity [J]. European Journal of Immunology. 2012;42(9):2263-2273.
[5] Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells [J]. Nature. 2006;441(7090):235-238.
[6] Zhou L, Lopes JE, Chong MM, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing ROR gamma t function [J]. Nature. 2008;453(7192):236-240.
[7] Monteleone G, Pallone F, Macdonald TT. Interleukin-21: a critical regulator of the balance between effector and regulatory T-cell responses [J]. Trends in Immunology. 2008;29(6):288-294.
[8] Chung Y, Chang SH, Martinez GJ, et al. Critical Regulation of Early Th17 Cell Differentiation by Interleukin-1 Signaling [J]. Immunity. 2009;30(4):576-587.
[9] Wei L, Laurence A, Elias KM, et al. IL-21 Is Produced by Th17 Cells and Drives IL-17 Production in a STAT3-dependent Manner [J]. Journal of Biological Chemistry. 2007;282(48):34605-34610.
[10] Mathur AN, Chang HC, Zisoulis DG, et al. Stat3 and Stat4 Direct Development of IL-17-Secreting Th Cells [J]. The Journal of Immunology. 2007;178(8):4901-4907.
[11] Brustle A, Heink S, Huber M, et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4 [J]. Nature Immunology. 2007;8(9):958-966.
[12] Ichiyama K, Yoshida H, Wakabayashi Y, et al. Foxp3 inhibits ROR gamma t-mediated IL-17A mRNA transcription through direct interaction with RORgammat [J]. Journal of Biological Chemistry. 2008;283(25):17003-17008.
[13] Stumhofer JS, Laurence A, Wilson EH, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system [J]. Nature Immunology. 2006;7(9):937-945.
[14] Laurence A, Tato CM, Davidson TS, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation [J]. Nature Immunology. 2007;8(9):903-912.
[15] Yang XO, Panopoulos AD, Nurieva R, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells [J]. Journal of Biological Chemistry. 2007;282(13):9358-9363.
[16] Moisan J, Grenningloh R, Bettelli E, et al. Ets-1 is a negative regulator of Th17 differentiation [J]. Journal of Experimental Medicine. 2007;204(12):2825-2835.
[17] Kolls JK, Linden A. Interleukin-17 family members and inflammation [J]. Immunity. 2004;21(4):467-476.
[18] Leipe J, Grunke M, Dechant C, et al. Role of Th17 cells in human autoimmune arthritis [J]. Arthritis & Rheumatism. 2014;62(10):2876-2885.
[19] Iwahashi M, Nakamura M, Katsuda M, et al. Tumor-infiltrating CD4+ Th17 cells produce IL-17 in tumor microenvironment and promote tumor progression in human gastric cancer [J]. Oncology Reports. 2011;25(5):1379-1385.
[20] Numasaki M, Fukushi JI, Ono M, et al. Interleukin-17 promotes angiogenesis and tumor growth [J]. Blood. 2003;101(7):2620-2627.
[21] Yang LJ, Qi YX, Hu J, et al. Expression of Th17 Cells in Breast Cancer Tissue and Its Association with Clinical Parameters [J]. Cell Biochemistry & Biophysics. 2012;62(1):153-159.
[22] Korn T, Bettelli E, Oukka M, et al. IL-17 and Th17 Cells [J]. Annual Review of Immunology. 2009;27:485-517.
[23] Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells [J]. Cell. 2006;126(6):1121-1133.
[24] McGeachy MJ, Chen Y, Tato CM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo [J]. Nature Immunology. 2009;10(3):314-324.
[25] Gaffen SL. Structure and signalling in the IL-17 receptor family [J]. Nature Reviews Immunology. 2009;9(8):556-567.
[26] Bedoya, S. K., Lam, B., Lau, K., & Joseph Larkin, I. (2013). Th17 Cells in Immunity and Autoimmunity [J]. Clinical and Developmental Immunology, 2013, 986789.
[27] Awasthi A, Kuchroo VK. Th17 cells: from precursors to players in inflammation and infection [J]. International Immunology. 2009;21(5):489-498.
[28] Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy [J]. Nature Reviews Immunology. 2010;10(4):247-262.
[29] Chi, X., Jin, W., et al. (2022). RORγt expression in mature TH17 cells safeguards their lineage specification by inhibiting conversion to TH2 cells [J]. Science Advances, 8(34), eabn7774.
[30] Kumar, R., Theiss, A. L., & Venuprasad, K. (2021). RORγt protein modifications and IL-17-mediated inflammation [J]. Trends in Immunology, 42(11), 1037.
[31] Ciofani, M., Madar, A., et al. (2012). A validated regulatory network for Th17 cell specification [J]. Cell, 151(2), 289.
[32] Capone, A., & Volpe, E. (2020). Transcriptional Regulators of T Helper 17 Cell Differentiation in Health and Autoimmune Diseases [J]. Frontiers in Immunology, 11, 348.
[33] Ziegler, S. F., & Buckner, J. H. (2009). FOXP3 and the Regulation of Treg/Th17 Differentiation [J]. Microbes and Infection / Institut Pasteur, 11(5), 594.
[34] Wang, J., Zhao, X., & Wan, Y. Y. (2023). Intricacies of TGF-β signaling in Treg and Th17 cell biology [J]. Cellular and Molecular Immunology, 20(9), 1002.
[35] Chang KK, Liu LB, et al. IL-27 triggers IL-10 production in Th17 cells via a c-Maf/RORγt/Blimp-1 signal to promote the progression of endometriosis [J]. Cell Death Dis. 2017 Mar 16;8(3):e2666.
[36] Murugaiyan, G., Mittal, A., et al. (2009). IL-27 Is a Key Regulator of IL-10 and IL-17 Production by Human CD4+ T Cells [J]. Journal of Immunology (Baltimore, Md. : 1950),183(4),2435.
[37] Park, E., & Ciofani, M. (2025). Th17 cell pathogenicity in autoimmune disease [J]. Experimental & Molecular Medicine, 57(9), 1913.
[38] Fitch, E., Harper, E., Skorcheva, I., Kurtz, S. E., & Blauvelt, A. (2007). Pathophysiology of Psoriasis: Recent Advances on IL-23 and Th17 Cytokines [J]. Current Rheumatology Reports, 9(6), 461.
[39] Park, E., & Ciofani, M. (2025). Th17 cell pathogenicity in autoimmune disease [J]. Experimental & Molecular Medicine, 57(9), 1913.
[40] Shen, W., & Durum, S. K. (2009). Synergy of IL-23 and Th17 Cytokines: New Light on Inflammatory Bowel Disease [J]. Neurochemical Research, 35(6), 940.
[41] AlMutairi, N., & Eassa, B. I. (2020). Comparing the efficacy and safety of IL-17 inhibitors for treatment of moderate-to-severe psoriasis: A randomized double blind pilot study with a review of literature [J]. Advances in Dermatology and Allergology/Postȩpy Dermatologii i Alergologii, 38(2), 281.


Comments
Leave a Comment