Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Hepatocellular carcinoma (HCC) is the third most common death-related cancer in the world and the sixth most common tumor. The major risk factors for liver cancer include chronic viral hepatitis, alcoholism, carcinogens or genetic diseases. Advances in cancer biology have shown that HCC development is associated with changes in several important cellular signaling pathways. Targeting these pathways can help prevent, delay or even reverse tumor development, which is of great significance for the treatment of hepatocellular carcinoma.
The present situation is that most patients (approximately 80%) diagnosed with hepatocellular carcinoma are at advanced stage and cannot be surgically resected due to the absence of early HCC symptoms, coupled with a lack of awareness of screening and unclear definition of screening strategies [1] [2]. Sorafenib, a first-line systemic therapy for advanced HCC, is a multikinase inhibitor. As the first effective drug to target liver cancer, it significantly prolongs patient survival and progression time, but its overall survival benefit is limited [3]. The lack of effective treatment options is one of the main reasons for the high mortality of HCC patients, which highlights the need for new treatments.
Several major molecular signaling pathways involved in the pathogenesis of liver cancer:
Receptors for the receptor tyrosine kinase pathway include the EGF receptor, the FGF receptor, the HGF receptor c-MET, the stem cell growth factor receptor c-KIT, the PDGF receptor, and the vascular endothelial growth factor (VEGF) receptor. These receptors activate multiple downstream signals. The Ras/MAPK and PI3K/Akt kinase signaling pathways are activated by ligand binding and phosphorylation of several growth factor tyrosine kinase receptors, including activation of the Grb2/Shc/SOS adaptor complex and downstream activation of Ras/Raf/Erk1/2 MAPK pathway, thereby activating AP-1 transcriptional activator.
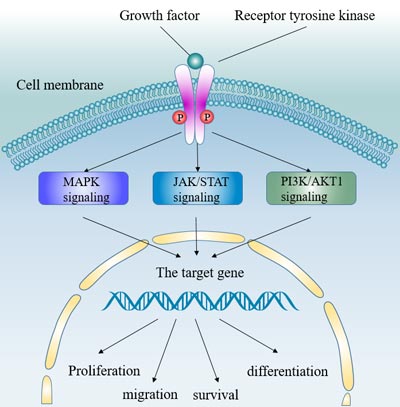
Figure 1 The receptor tyrosine kinase signaling pathway
In hepatocellular carcinoma, many growth factors, including VEGF receptor, EGFR, IGF and HGF/c-MET, have significant effects on the occurrence and pathogenesis of tumors.
Tumor growth and invasion of liver cancer are dependent on angiogenesis disorders. VEGF is a key angiogenic factor. Therefore, VEGF plays an important regulatory role in liver cancer. Studies have shown that both VEGF and VEGFR expression are increased in HCC patients (including VEGFR-1, VEGFR-2, and VEGFR-3). Hepatitis b x antigen is associated with the up-regulation of VEGFR-3 [4]. VEGF expression is induced by hypoxic tumor environment (HIF-2), EGFR activation and COX-2 signaling [5] [6].
High expression of VEGF was associated with HCC tumor grade [7], poor prognosis after resection, disease recurrence, vascular invasion and portal vein embolism Therefore, VEGF growth factor can be used as a therapeutic target for HCC.
Sorafenib, a drug currently used to treat hepatocellular carcinoma, has major molecular targets including vascular endothelial factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR) and Raf [8].This indicates that targeted growth factor is an effective treatment.
EGF is a ligand that binds to EGFR and plays an important role in tumor angiogenesis and proliferation. These effects are mainly achieved by activating the RAF/MEK/ERK and PI3K/AKT/mTOR pathways. Studies have shown that EGFR signaling plays a role in early liver cancer. In HCC, EGF and related growth factors are generally up-regulated.
The IGF signaling pathway regulates a variety of cellular processes, including proliferation, motility, and apoptosis inhibition. The IGF signaling pathway is activated by binding of ligands (IGF-1 and IGF-2) to membrane-bound receptors (IGF-1R).
Major processes in the IGF signaling pathway: ligand binding to IGF-1R initiates autophosphorylation of the receptor, followed by phosphorylation of intracellular targets, which in turn activates downstream cellular effector factors and ultimately activates the PI3K, protein kinase B, and RAF/MEK/ERK pathways.
In hepatocellular carcinoma, the insulin-like growth factor (IGF) signaling pathway is frequently dysregulated. The imbalance of IGF signal is mainly manifested in the level of IGF-2 bioavailability. IGF-2R is overexpressed in most hepatocellular carcinomas, and this excess ligand availability leads to increased binding to the receptor and further action on the MAPK and PI3K/AKT/mTOR pathways.
HGF is a cytokine involved in the invasion of malignant tumors. It works primarily by binding to the tyrosine kinase receptor c-MET. Major functions of c-MET include tissue regeneration, cell proliferation, migration, survival, branch morphogenesis, and angiogenesis. Binding of HGF results in homodimerization and self-phosphorylation of the c-MET receptor and further phosphorylation of adaptor proteins (including GRB2) and GAB1 (GRB2-associated -binding protein 1), which then activate downstream effector molecules (including phospholipase C, PI3K, and ERK). C-MET phosphorylation can also be activated by the epidermal growth factor receptor (EGFR), cell adhesion, or replacement ligand des-γ-carboxythrombin.
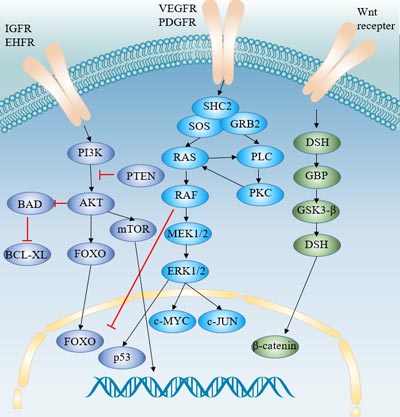
Figure 2 Signaling pathways involved in the pathogenesis of hepatocellular carcinoma
RAF/MEK/ERK pathway is a ubiquitous signal transduction pathway whose main role is to regulate cell proliferation, differentiation, angiogenesis and survival. Activation of this pathway contributes to tumor development, progression and metastasis.
The ERK/MAPK pathway is regulated by various growth factors, and the binding of growth factors leads to the phosphorylation of receptors and the activation of an adaptive molecular complex. This in turn activates the RAF/MEK/ERK pathway, triggering a series of specific phosphorylation events [5]. The intermediate signaling pathway is regulated by mitogen/extracellular protein kinases MEK1 and MEK2, which are responsible for phosphorylation of downstream signaling molecules ERK1 and ERK2. ERK1/2 regulates cellular activity through a variety of substrates that act on the cytoplasm and nucleus.
In hepatocellular carcinoma, RAF/MEK/ERK pathway is usually activated by two main mechanisms:
It is reported that RAF/MEK/ERK pathway also can be activated by HBV infection in HCC.
The role of RAF/ERK/MAPK pathway in tumorigenesis indicates that this pathway is also an effective therapeutic target for HCC.
Activation of the PI3K/AKT/mTOR signaling pathway is a major determinant of tumor cell growth and survival in multiple solid tumors. mTOR pathway plays an important role in the pathogenesis of liver cancer, with 15% ~ 41% of liver cancer patients suffering from mTOR pathway aberration.
In the PI3K/AKT/mTOR signaling pathway, the binding of growth factors (IGF and EGF) to receptors activates PI3K. PI3K then produces a second lipid messenger, PIP3b (phosphoinositol triphosphate), which in turn activates the serine/threonine kinase AKT. Activated AKT regulates various transcription factors and phosphorylates several cytoplasmic proteins, including BAD and mTOR. The mTOR protein in turn regulates phosphorylation of the p70-S6 kinase, the serine threonine kinase, and the translational inhibitory protein PHAS-1/4E-BP. These proteins regulate the translation of cell cycle regulatory proteins and promote cell cycle progression.
mTORC1 and mTORC2 are important components in this signaling pathway. mTORC1 is a downstream signal of AKT, which activates S6 kinase to regulate protein synthesis and induces cell cycle progression from G1 phase to S phase [9].
In hepatocellular carcinoma, the overactive EGF and IGF signaling pathways are responsible for inducing the PI3K/AKT/mTOR pathway and promoting tumor progression. In addition, PTEN dysfunction is also the cause of excessive activation of PI3K/AKT/ mTOR pathway [10]. PTEN inhibits this pathway by reversing the PI3K response and blocking Akt activation. In hepatocellular carcinoma, PTEN gene expression is down-regulated, and its down-regulation is regulated by HBX protein. The interaction between GPC3 and FGF-2 is frequently observed in liver cancer cells and is responsible for phosphorylation of ERK and AKT [11].
There is also some evidence that the PI3K/AKT/mTOR signal may be activated by somatic mutations in the PI3K-catalyzed gene PIK3CA.
The PI3K/ AKT/mTOR pathway plays a key role in the pathogenesis of HCC. As a key role in mTOR signaling pathway, mTOR activity is often increased in HCC. Inhibition of mTOR activity can reduce cell proliferation in vitro and tumor growth in xenograft mouse models [12]. Blocking mTOR with everolimus slows tumor growth and improves survival. HSP90 is involved in the folding and activity of many oncoprotein proteins. Therefore, blocking HSP90 blocks the rapamycin-induced AKT signaling pathway, thereby enhancing the anti-tumor effect of mTOR inhibitor rapamycin [13].
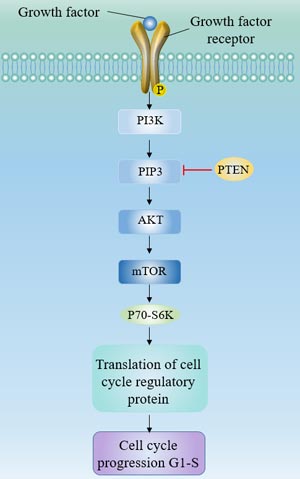
Figure 3 The PI3Kinase/AKT/mTOR pathway
β-catenin in the Wnt signaling pathway is an important factor in the development of early and early carcinogenic events in HCC.
Wnt is a glycoprotein ligand that acts as a ligand for the Frizzled cell surface receptor family and activates a receptor-mediated signaling pathway [14].
In the absence of Wnt ligands, β-catenin binds to E-cadherin in most cells. Cytosol β-catenin forms complexes with adenomatous polyposis coli (APC) and AXIN1 or AXIN2, which mediates casein kinase 1 and glycogen synthase kinase 3b (GSK3b), resulting in continuous β-catenin Phosphorylation and degradation.
In the presence of Wnt ligand, extracellular Wnt ligand binds to the cell surface Frizzled family receptor, resulting in phosphorylation and inhibition of GSK3b, leading to accumulation of cytosolic β-catenin. Then β-catenin is translocated to the nucleus, interacts with TCF and LEF transcription factors, and activates transcription of target genes, including cyclin D1, c-Myc, c-MET, FGF4, metalloproteinases, and VEGF. Accumulation of β-catenin provides growth advantages for tumor cells by promoting proliferation and inhibiting differentiation.
In hepatocellular carcinoma, aberrant activation of the Wnt pathway is primarily a mutation in β-catenin. Studies have shown that treatment of HCC cell lines with TGF-β results in activation of β-catenin, suggesting that this pathway may also be mediated by TGF-β.
Glypican-3 (GPC3) is a cell surface protein. The expression level of GPC3 is higher in HCC cell lines. GPC3 exerts its carcinogenic effect by activating Wnt/β-catenin signaling.
Cadherin 17 (CDH17) is an up-regulated adhesion molecule in HCC, which is related to tumorigenesis in various regions of the gastrointestinal tract [15]. Inhibition of CDH17 leads to the relocation of nuclear β-catenin to the cytoplasm, thereby weakening the Wnt/β-catentin signaling pathway [16]. Targeting CDH17 simultaneously inactivates the Wnt/β-catenin signal pathway.
The role of Wnt in the regulation of liver regeneration and the maintenance and self-renewal of pluripotent stem and progenitor cells suggests that it may be an ideal target for cancer therapy.
Different targeting targets can be proposed for the different activation of the Wnt channel:
Several drugs currently in clinical use have been shown to have anti-Wnt pathway activity. These drugs include non-selective cyclooxygenase (COX) [17], inhibitors indomethacin, sulinic acid, aspirin, and nitric oxide releasing aspirin, and selective inhibitors celecoxib and rofekoxib.
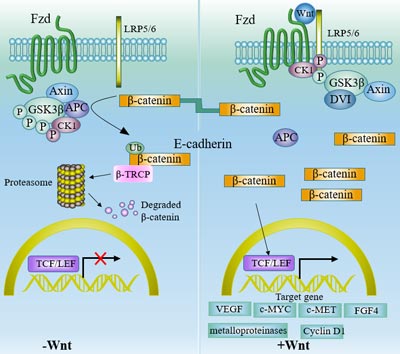
Figure 4 Wnt/β-catenin Pathway
The ubiquitin-proteasome system is a highly conserved cellular protein degradation system in eukaryotic systems. The ubiquitin-proteasome system plays an important role in maintaining cell homeostasis such as cell cycle regulation, apoptosis, receptor signal transduction and endocytosis.
The ubiquitin-labeled protein is degraded by the proteasome or absorbed by endocytosis and degraded in the lysosome. Ubiquitin-proteasome-mediated protein degradation is accomplished through a series of steps including ubiquitin-activating enzyme (E1), polyubiquitin-coupled enzyme (E2s), and ubiquitin ligase (E3s).
Deregulation of NF-κB in HCC is one of the carcinogenic events induced by ubiquitin-proteasome system defects. Furthermore, HBV-expressed proteins [18] and HCV-expressed proteins [19] cause changes in the ubiquitin-proteasome system, leading to viral replication, liver tumorigenesis and host immune damage. This further illustrates the importance of the ubiquitin-proteasome system in the development of HCC.
A variety of cancer-associated proteins are regulated by the ubiquitin-proteasome system, including:
Some cancer-related proteins are also involved in the ubiquitin conjugation mechanism: FRA6E fragile locus protein Parkin, an E3 ligase targeting cyclin E and P38, have been shown to act as tumor suppressors in HCC.
Hedgehog (Hh) signaling pathway is a highly conserved system that plays a crucial role in tissue pattern, cell differentiation and proliferation [20]. Abnormal activation of Hh signaling pathway leads to the occurrence and development of tumorigenesis in pancreas, colon, stomach [21], lung [22], prostate [23], breast, skin and other cancers.
Recent studies have shown that the Hh pathway is abnormally activated in human HCC. GPC3 interacts with the hedgehog signaling pathway in regulating developmental growth [24]. And the GPC3-hedgehog signaling pathway is thought to contribute to the development of HCC. The Hh pathway plays an important role in the development and invasion of HCC. Blocking the Hh signaling pathway may be a potential target for new HCC treatment strategies [25]. Downregulation of Gli2 with a ligand blocking antibody or KAAD-cyclopamine (selective Smo antagonist) or inhibition of this pathway has been shown to delay tumor growth and reduce tumor size.
Ras and Jak/Stat pathways are generally activated in HCC, and Ras pathway is the main signal network promoting cell proliferation and survival. The binding of different growth factors (eg EGF and IGF-1) to the receptor (eg EGFR, IGF-1R) induces the activation of Ras, which in turn activates c-RAF, MEK and ERK.
Ras and Jak/Stat inhibitors and demethylating agents can be used as a treatment for human liver cancer.
References
[1] Bruix J, Sherman M. Management of hepatocellular carcinoma: An update [J]. Hepatology, 2011, 53(3): 1020-1022.
[2] X-P. Chen, F-Z. Qiu, Z-D. Wu, et al. Long-term outcome of resection of large hepatocellular carcinoma [J]. Br J Surg, 2010, 93(5): 600-606.
[3] Schwartz J D. Sorafenib in Advanced Hepatocellular Carcinoma [J]. N Engl J Med, 2008, 359(23): 378-390.
[4] Lian Z, Liu J, Wu M, et al. Hepatitis B x antigen up-regulates vascular endothelial growth factor receptor 3 in hepatocarcinogenesis [J]. Hepatology, 2007, 45: 1390–1399.
[5] Avila MA, Berasain C, Sangro B, et al. New therapies for hepatocellular carcinoma [J]. Oncogene, 2006, 25: 3866–3884.
[6] Bangoura G, Liu ZS, Qian Q, et al. Prognostic significance of HIF-2 alpha/EPAS1 expression in hepatocellular carcinoma [J]. World J Gastroenterol, 2007, 13: 3176–3182.
[7] Yamaguchi R, Yano H, Iemura A, et al. Expression of vascular endothelial growth factor in human hepatocellular carcinoma [J]. Hepatology, 1998, 28: 68–77.
[8] Wilhelm S M, Carter C, Tang L Y, et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis [J]. Cancer Research, 2004, 64(19): 7099-7109.
[9] Bjornsti M A, Houghton P J. The TOR pathway: a target for cancer therapy [J]. Nature Reviews Cancer, 2004, 4(5): 335-48.
[10] Hu T H, Huang C C, Lin P R, et al. Expression and prognostic role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular carcinoma [J]. Cancer, 2010, 97(8): 1929-1940.
[11] Midorikawa Y, Ishikawa S, Iwanari H, et al. Glypican-3, overexpressed in hepatocellular carcinoma, modulates FGF2 and BMP-7 signaling [J]. International Journal of Cancer, 2003, 103(4): 455-465.
[12] Villanueva A, Chiang D Y, Newell P, et al. Pivotal Role of mTOR Signaling in Hepatocellular Carcinoma [J]. Gastroenterology, 2008, 135(6): 1972-1983.e11.
[13] Lang S A, Moser C, Fichnterfeigl S, et al. Targeting heat-shock protein 90 improves efficacy of rapamycin in a model of hepatocellular carcinoma in mice [J]. Hepatology, 2010, 49(2): 523-532.
[14] Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors [J]. ONCOGENE, 2006, 25(27): 3778-3786.
[15] Wang X Q, Luk J M, Leung P P, et al. Alternative mRNA splicing of liver intestine-cadherin in hepatocellular carcinoma [J]. Clinical Cancer Research, 2005, 11(1): 483-489.
[16] Liu L X, Lee N P, Chan V W, et al. Targeting cadherin-17 inactivates Wnt signaling and inhibits tumor growth in liver carcinoma [J]. Hepatology, 2009, 50(5): 1453-63.
[17] Lucrecia Márquez-Rosado, María Cristina Trejo-Solís, Claudia María García-Cuéllar, et al. Celecoxib, a cyclooxygenase-2 inhibitor, prevents induction of liver preneoplastic lesions in rats [J]. Journal of Hepatology, 2005, 43(4): 0-660.
[18] Hu Z, Zhang Z, Doo E, et al. Hepatitis B Virus X Protein Is both a Substrate and a Potential Inhibitor of the Proteasome Complex [J]. Journal of Virology, 1999, 73(9): 7231-7240.
[19] Munakata T, Nakamura M, Liang Y, et al. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase [J]. Proc Natl Acad Sci USA, 2005, 102(50): 18159-18164.
[20] Altaba A R I, Pilar Sánchez, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells [J]. Nature Reviews Cancer, 2002, 2(5): 361-72.
[21] Katoh Y, Katoh M. Hedgehog signaling pathway and gastric cancer [J]. Cancer Biology & Therapy, 2005, 4(10): 1050-1054.
[22] Velcheti V, Govindan R. Hedgehog Signaling Pathway and Lung Cancer [J]. Journal of Thoracic Oncology, 2007, 2(1): 7-10.
[23] Thiyagarajan S, Bhatia N, Reaganshaw S, et al. Role of GLI2 transcription factor in growth and tumorigenicity of prostate cells [J]. Cancer Research, 2007, 67(22): 10642-10646.
[24] Capurro M I, Xu P, Shi W, et al. Glypican-3 Inhibits Hedgehog Signaling during Development by Competing with Patched for Hedgehog Binding [J]. Developmental Cell, 2008, 14(5): 0-711.
[25] Tian. Role of Hedgehog signaling pathway in proliferation and invasiveness of hepatocellular carcinoma cells [J]. International Journal of Oncology, 2009, 34(3): 829-836.


Comments
Leave a Comment