Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
A cell is not only the basic unit of biological structure and function but also of organismal aging. Most cells of an organism go through several stages during their lives: proliferation, differentiation, senescence, and death. It is well known that nothing is permanent, and the ability of cells to proliferate is no exception. One barrier to the continuous division of the cultured cells is the senescence, which was first proposed to describe the limited prolife [1]. The number of times (about 50) that normal human embryonic cells can divide before they succumb trative competence of normal human fibroblasts in culture by Leonard Hayflick and Paul Moorhead in 1961 o senescence is known as the 'Hayflick Limit'.
In this review, we will introduce the definition of cellular senescence, characteristics of senescent cells, two pathways that regulate senescence growth arrest, as well as cellular senescence markers.
1. What Is Cellular Senescence?
2. Characteristics of Senescent Cells
Cellular senescence is a process in which proliferating cells terminate division and permanently exit from the cell cycle. Different from quiescence, which is reversible with appropriate mitogens, senescence is a permanent cell cycle halt state. Senescence occurs when cells encounter a variety of stimuli, including telomere dysfunction, DNA damage, mitochondrial dysfunction, oxidative stress, oncogene activation, nutrient signaling dysfunction, chronic inflammation, mitogenic signals, or exposure to exogenous toxins [2-4].
Cellular senescence exerts dual effects on cells. It plays beneficial roles in embryo development, maintenance of tissue homeostasis, wound healing, host immunity, and inhibition of tumor progression [5-8]. On the contrary, cell senescence is one of the major causes of aging and age-associated diseases. It promotes tissue regeneration during development and after injury, but impairs cells' regenerative and functional capacity, leading to inflammation and tumorigenesis in senescent organisms. Senescent cells steadily accumulate in late pathogenic phases due to inadequate clearance, contributing to the aggravation of pathological symptoms. Additionally, senescent cells can promote cancer progression by up-regulating and releasing factors that can change the tumor microenvironment, including inflammatory cytokines, chemokines, and growth regulators.
Senescent cells remain viable or metabolically active [9] [10] and show some obvious changes, with four typical characteristics, including irreversible cell cycle arrest, senescence-associated secretory phenotype (SASP), macromolecular damage, and deregulated metabolism. Senescent cells can also experience structural and morphological alterations, such as an enlarged, flattened, multinucleated morphology with increased vacuoles, modified plasma membrane composition, and an astonishing nuclear expansion.
Irreversible cell cycle arrest is one of the most basic characteristics of cell senescence. This phenomenon is one of the most basic and indispensable indicators for identifying cell senescence in vitro.
Senescent cells secrete numerous factors, including inflammatory cytokines, chemokines, growth regulators, angiogenic factors, and matrix metalloproteinases, collectively known as the SASP. SASP is another typical marker of senescent cells. These SASP factors cooperate with the immune system to improve the cell microenvironment, and affect the proliferation and differentiation of neighboring cells, resulting in bidirectional regulation of organ aging and the occurrence and development of tumors.
| SASP Types | Components |
|---|---|
| Interleukins (IL) | IL-1a, IL-1b, IL-6, IL-7, IL-13, IL-15 |
| Chemokines (CXCL, CCL) | IL-8, GRO-a, GRO-b, GRO-gamma, MCP-2, MCP-4, MIP-1a, MIP-3a, HCC-4, eotaxin-3, TECK, ENA-78, I-309, I-TAC |
| Other inflammatory factors | GM-CSE, G-CSE, IFN-γ, BLC, MIF |
| Growth factors and regulators | Amphiregulin, Epiregulin, Heregulin, EGF, bFGF, HGF, KGF (FGF7), VEGF, Angiogenin, SCF, SDF-1, PIGF, NGF, IGFBP-2, IGFBP-3, IGFBP-4, IGFBP-6, IGFBP-7 |
| Proteases and regulators | MMP-1, MMP-3, MMP-10, MMP-12, MMP-13, MMP-14, TIMP-1, TIMP-2, PAI-1, PAI-2, tPA, uPA, Cathepsin B |
| Soluble or shed receptors or ligands | ICAM-1, ICAM-3, OPG, sTNFRI, TRAIL-R3, Fas, sTNFRII, uPAR, SGP130, EGF-R |
| Nonprotein soluble factors | PGE2, Nitric oxide, Reactive oxygen species |
| Insoluble factors (ECM) | Fibronectin, Collagens, Laminin |
Table 1: Claasification of SASP factors (The information in this table is cited from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4166495/)
There are three kinds of macromolecular damage in senescent cells: DNA damage, protein damage, and lipid damage. Telomere shortening is closely associated with the cell cycle arrest. In senescent cells, mitochondrial dysfunction, ATP generation blockade, and massive ROS production eventually lead to protein damage and lipid damage.
The number and size of lysosomes increased in senescent cells. SA-B-GAL accumulates in lysosomes.
So far, no senescence marker or hallmark has been established that is completely unique to the senescent state. Furthermore, not all senescent cells express all of the senescence markers that are available. Some available senescence markers include short and dysfunctional telomeres, p16INK4A expression, persistent DNA damage response, the release of SASP, senescence-associated heterochromatin foci (SAHF), enhanced senescence-associated β-galactosidase (SA-β-gal) activity is one of the earliest biomarkers for the identification of senescent cells, and loss of Lamin B1.
| Different aspects | Senescent Features or Markers | Further elucidation |
|---|---|---|
| Morphologic characteristics | Increased size | Senescent cells usually appear a multinucleated, large, polygonal, and flattened morphology, and assume spindle and vacuolization features [11]. Golgi apparatus and lysosomes in senescent cells increases in number, and intracytoplasmic particles increase significantly. These morphologic features can be assessed using approaches such as brightfield microscopy and may reflect their altered metabolism and organelle homeostasis. |
| Increased granularity | ||
| Cell cycle arrest | p53 and phospho-p53 | Accumulation of phosphorylated p53 promotes the activation of cyclin dependent kinase inhibitors (CDKIs), eventually resulting in cell cycle arrest. |
| 53BP1 | 53BP1 is a mediator of cellular senescence by in p53. Defects in 53BP1-p53 interaction lead to inefficient p53-dependent cell cycle checkpoint activation, ultimately resulting in cellular senescence. | |
| P21 | When engagement to CDK2, p21 inhibits the cell cycle by preventing the G1-S transition. | |
| p16 | Staining for p16INK4A is one of the best-acknowledged markers for senescence [12]. Most senescent cells express p16INK4a. p16 binds to CDK4 and CDK6, leading to the cell cycle arrest at the G1 phase. | |
| Rb and phospho-Rb | Phosphorylation of Rb promotes the progression of the cell cycle, and phospho-Rb is not detected in senescent cells. Inhibition of the cell-cycle by various CDKIs, including p21 and p16, leads to hyperactivation of Rb; this ultimately promotes the arrest of the cell cycle and senescence. | |
| Mitochondrial changes | Increased size/number | Senescent cells have increased mitochondrial abundance, as well as changed membrane potential, increased ROS generation, oxidative phosphorylation, and oxygen consumption. They can be used to enhance senescent cell phenotyping. |
| Increased ROS production | ||
| Decreases membrane integrity | ||
| Lysosomal changes | Increased size/number | Lysosomal abundance in senescent cells allows for senolysis through enhanced autophagy. |
| Enhanced SA-β-gal activity | SA-β-gal activity is one of the earliest biomarkers for the identification of senescent cells and can be detected at pH 6.0 [13]. SA-β-gal partly reflects the increase in lysosomal mass [13] [14]. It accumulates in the lysosome in senescent cells. | |
| Lipofuscin accumulation | Lipofuscins are aggregates of lysosomal byproducts and accumulate within senescent cells. | |
| Nuclear changes | Telomere shortening | It is the major cause for aging [15] and is one of the first and most prominent characteristic mechanisms of cellular senescence. |
| telomere-associated foci | These markers all reflect DDR. H2AX is phosphorylated at Ser139 and becomes γ-H2AX in response to DNA damage, which makes it a potent tool for research on the DDR pathway and senescence. | |
| SAHF | ||
| γ-H2AX | ||
| Lamin B1/LMNB1 loss | Lamin B1 is involved in structural changes of the nucleus with senescence [16] and is lost in senescent cells. | |
| Decreased DNA replication | Senescent cells proliferation is halted and retain low replication capacity. | |
| Ki67 | Ki67 is a nuclear protein frequently used as a marker for proliferating cells. Since senescent cells permanently exit from the cell cycle, so they do not express Ki67. | |
| Additional selected features | Cytosolic DNA/cGAS–STING activation | SASP includes interleukins like IL-6, chemokines like IL-8, and growth factors like VEGF7 (Refer to table 1). They affect neighbouring tissues and may induce senescence in adjacent cells. |
| LINE-1 retrotransposon de-repression | ||
| remodeling of SASP-associated super-enhancers |
Table 2: Senescent markers and features. (The information in this table comes from: https://www.nature.com/articles/s43587-021-00121-8)
In senescent cells, DNA and other types of macromolecular damage ultimately results in proliferation cessation by activating the p53/p21CIP1 and p16INK4a/RB tumor suppressor pathways. These two pathways are regulated by the gatekeeper tumor suppressor proteins p53 and pRb [17] [18].
Following being stimulated by numerous stress signals such as telomere erosion, DNA damage, and oncogene activation, p53 is activated by two posttranslational modifications: phosphorylation (ATM-Chk2 or ATR-Chk1) and acetylation (p300/CBP, PCAF, Tip60/hMOF, and SIRT1), and by enhances protein stability (ARF-MDM2), and by increased translation rates (RNPC1, HuR, and RPL26). And then, active p53 activates the expression of pro-senescent targets such as p21 and E2F7, which contributes to G1 cell cycle arrest and mitotic genes suppression, respectively, resulting in cellular senescence or other cellular responses [19] [20].
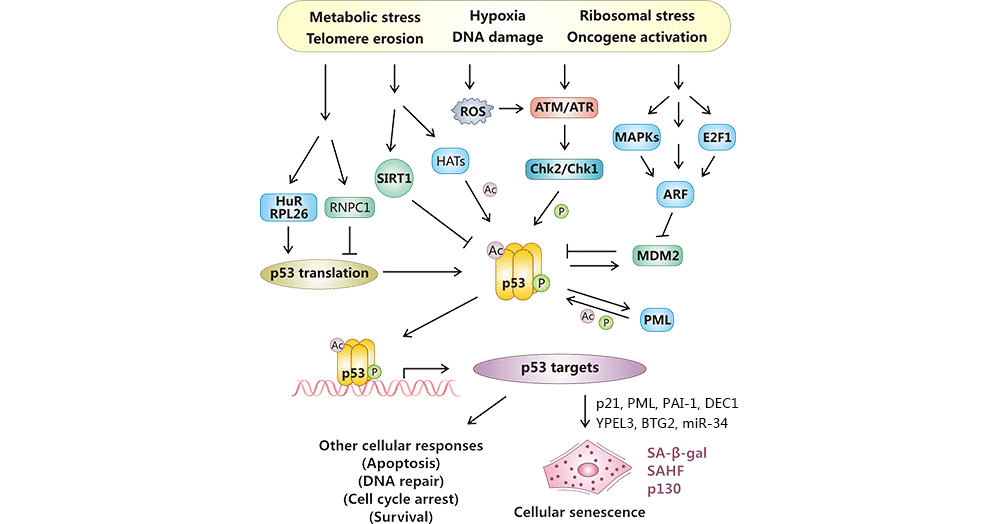
Figure 1: Cellular senescence regulation by p53.
The picture is cited from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3784259/
Various internal or external stress factors trigger the DNA-damage response (DDR) pathway, which triggers p16INK4A pathways. p16INK4A inactivates Cdk4 and Cdk6, results in accumulation of phosphorylated pRb, inhibits the expression of E2F transcription factors, and promotes cell cycle arrest or senescence [21].
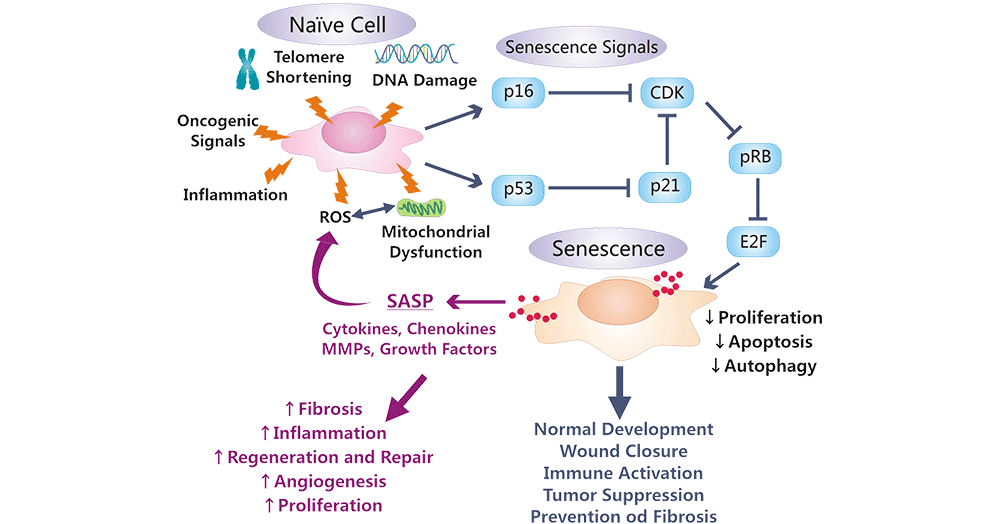
Figure 2: p53- and pRB-mediated regulation, effects, and functional and morphological alterations occur in a senescent cell.
The picture is cited from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6589594/
CUSABIO also provides some antibodies that will help to recognize and identify frequently used cellular senescence markers.
| Target Molecules | Function | Antibody Products |
|---|---|---|
| phospho-Histone H2AX (S139) | DNA damage response (DDR) markers | Recombinant Phospho-Histone H2AX (S139) Antibody |
| P16INK4A | Tumor suppressors / cell cycle regulators | CDKN2A Antibody |
| P21 | Tumor suppressors / cell cycle regulators | CDKN1A Antibody |
| P53 | Tumor suppressors / cell cycle regulators | Recombinant TP53 Antibody |
| Phospho-RB1 (S780) | Tumor suppressors / cell cycle regulators | Recombinant Phospho-RB1 (S780) Antibody |
| Ki67 | Proliferation marker | MKI67 Monoclonal Antibody |
| Beta-galactosidase | Lysosome-associated protein; activity enhances | lacZ Monoclonal Antibody |
| IL-6 | SASP maker; the most prominent cytokine of the SASP; significantly increases | Recombinant IL-6 Antibody |
| IL-8 | SASP maker; most senescent cells overexpress IL-8 | CXCL8 Monoclonal Antibody |
| Lamin B1 | Nuclear lamina markers; absent in senescent cells | LMNB1 Monoclonal Antibody |
References
[1] Hayflick, P.S. Moorhead. The serial cultivation of human diploid cell strains [J]. Exp. Cell Res., 25 (1961), pp. 585-621.
[2] B.G. Childs, M. Durik, et al. Cellular senescence in aging and age-related disease: from mechanisms to therapy [J]. Nat. Med., 21 (2015), pp. 1424-1435.
[3] D. Muñoz-Espín, M. Serrano. Cellular senescence: from physiology to pathology [J]. Nat. Rev. Mol. Cell Biol., 15 (2014), pp. 482-496.
[4] J.M. van Deursen. The role of senescent cells in ageing [J]. Nature, 509 (2014), pp. 439-446.
[5] Kuwano K, Araya J, et al. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF) [J]. Respir Investig. 54:397–406. 2016.
[6] Shimizu H, Bolati D, et al. NF-κB plays an important role in indoxyl sulfate-induced cellular senescence, fibrotic gene expression, and inhibition of proliferation in proximal tubular cells [J]. Am J Physiol Cell Physio. 2011 301:C1201–C1212.
[7] Lansu K and Gentile S. Potassium channel activation inhibits proliferation of breast cancer cells by activating a senescence program [J]. Cell Death Dis. 2013 Jun; 4(6): e652.
[8] Ma J, Hu X, Liao C, et al. Gypenoside L inhibits proliferation of liver and esophageal cancer cells by inducing senescence [J]. Molecules. 2019, 24(6):1054.
[9] J.R. Dörr, Y. Yu, M. Milanovic, et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy [J]. Nature, 501 (2013), pp. 421-425.
[10] Matsumura T, Zerrudo Z, Hayflick L. Senescent human diploid cells in culture: survival, DNA synthesis and morphology [J]. J. Gerontol. 1979;34:328–334.
[11] Kuilman T, Michaloglou C, et al. The essence of senescence [J]. Genes Dev 2010; 24: 2463–2479.
[12] Krishnamurthy J, Torrice C, et al. Ink4a/Arf expression is a biomarker of aging [J]. J Clin Invest (2004) 114:1299–307.
[13] Dimri G.P., Lee X., et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo [J]. Proc. Natl. Acad. Sci. USA. 92:9363–9367.
[14] Lee B.Y., Han J.A., et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase [J]. Aging Cell. 2006. 5:187–195.
[15] Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts [J]. Nature (1990) 345:458–60.
[16] Freund A, Laberge R-M, et al. Lamin B1 loss is a senescence-associated biomarker [J]. Mol Biol Cell (2012) 23:2066–75.
[17] F. Bringold and M. Serrano. Tumor suppressors and oncogenes in cellular senescence [J]. Exp. Gerontol., 35 (2000), pp. 317-329.
[18] A.S. Lundberg, W.C. Hahn, et al. Genes involved in senescence and immortalization [J]. Curr. Opin. Cell Biol., 12 (2000), pp. 705-709.
[19] Rufini, A., Tucci, P., et al. Senescence and aging: the critical roles of p53 [J]. Oncogene 32, 5129–5143 (2013).
[20] Qian Y and Chen X. Senescence regulation by the p53 protein family [J]. Methods Mol Biol. 2013;965:37-61.
[21] Parikh P, Wicher S, et al. Cellular senescence in the lung across the age spectrum [J]. Am J Physiol Lung Cell Mol Physiol. 2019;316(5):L826-L842.


Comments
Leave a Comment