Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
As you know, cytokines provide cells with the ability to communicate with one another and exert complex multicellular behavior. Neuroinflammation is a common feature of neurological disorders in the brain, including Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), Parkinson's disease and Huntington's, etc. It is a complex response to brain injury, and often companied by the release of proinflammatory cytokines and chemokines. This review focus on the role of pro-inflammatory cytokines produced by neuroinflammation.
Before starting to introduce the pro-inflammatory cytokine produced by neuroinflammation, we discuss the sources of neuroinflammation. Neuroinflammation comprises biochemical and cellular responses of the nervous system to injury, infection or neurodegenerative diseases. Generally speaking, as the Figure 1 shows, aging, metabolic diseases and viral infections are the primary sources of inflammation that can affect vessels and neurons, leading to neurodegeneration. SVD, small vessel disease [1].
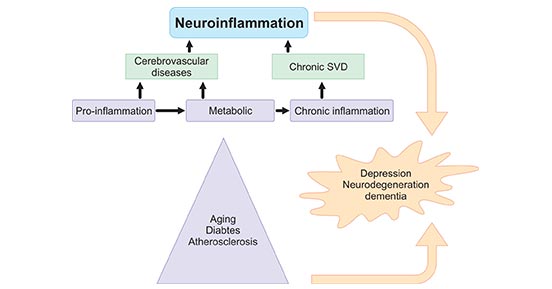
Figure 1. Sources of neuroinflammation
*This diagram is derived from reference 1. [1]
The cellular and molecular mechanisms of neuroinflammation are likely the same in aging and metabolic diseases such as hypertension, diabetes, depression, dementia or after cerebral insult [2], and are considered as silent contributors of neuroinflammation. Take the elderly as an example, inflammatory mechanisms are associated with the pathogenesis of dementia and functional impairment. Systemic and local CNS inflammation significantly leads to cerebral small vessel disease (SVD)-vascular dementia [3] [4], hypothesized as microvascular changes that result in a state of chronic hypoperfusion, contributing to continuous oligodendrocyte death and the consecutive degeneration of myelinated fibers that increase low-grade inflammation amplification of the risk of stroke [5].
Classically defined neuroinflammatory conditions, as observed in inflammatory demyelinating diseases (for example, multiple sclerosis) and infections (bacterial and viral encephalitis), are characterized by leukocytes invading the CNS parenchyma and a drastic loss of blood–brain barrier (BBB) integrity. Here, lymphocytes and myeloid cells are the main mediators of tissue damage and deliver cytokines to the tissue, fuelling the inflammatory cascade. Recent studies have revealed that many proinflammatory cytokines released in neuroinflammation are key players in neuroinflammation mediated by invading leukocytes, involving IL-23, IL-6, IL-1β, IFNγ, TNF, and GM-CSF [6] [7] [8]. IL-1β and IL-6 can be sensed by all cells in the inflamed CNS and are central to the inflammatory process. IL-23 is sensed by T cells, fuelling their pathogenic signature. Granulocye–macrophage colony-stimulating factor (GM-CSF) in turn licenses inflammatory monocyte-derived cells to cause tissue damage [9].
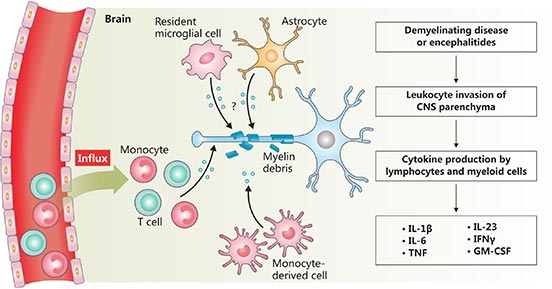
Figure 2. The diagram of neuroinflammation
*This diagram is derived from reference 9.
IL-23, a heterodimeric cytokine is composed of the p19 subunit unique to IL-23 and the p40 subunit common to IL-12. It is primarily produced by antigen-presenting cells (APCs) in secondary lymphoid tissues such as macrophages, dendritic cells, and microglia. IL-23 serves to polarize differentiating T cells toward different effector function phenotypes, such as IL-17-producing T helper (Th17) cells [10]. Accumulating studies have demonstrated that the IL-23/Th17 axis is thought to play a critical role in neuroinflammation [11].
Blockade of IL-23 ameliorates experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis (MS), which is an inflammatory demyelinating disease of the central nervous system [12]. It is generally accepted that MS begins with the formation of acute inflammatory lesions that were mediated by autoreactive T cells and B cells because of the breakdown of the blood–brain barrier (BBB). The demyelinating plaques were dominated by activated T cells and macrophages associated with oligodendrocyte destruction. YuhongYang et al. have reported that IL-23-driven Th-17 cells is believed to contribute significantly to the pathogenesis of MS and EAE [13]. Moreover, Li et al. reported that activated macrophages/microglia produce IL-23 p19 in active and chronic active multiple sclerosis lesions [14].
As mentioned before, neuroinflammation is commonly seen in the postmortem brains of AD patients [15]. Accumulation of β-amyloid (Aβ) remained the hallmark in AD pathology. Many researches have confirmed that Aβ can induce expression of IL-6 in astrocytes and microglia in culture [16]. In hippocampal neurons, Aβ and IL-6 were both able to induce synaptic dysfunction [17].
AD is accompanied by a rise in IL-6 levels in the very early stage of AD, but it cannot be used as a biomarker because it is also increased with mere age. Both astrocytes and microglia can produce this cytokine and, at the same time, IL-6 induces their proliferation and activation and enhances the production and release of inflammatory mediators: prostaglandins, cytokines, chemokines and acute phase proteins, like APP. Moreover, IL-6 upregulates the cdk5/p35 complex which is involved in the hyperphosphorilation of Tau protein.
IL-1β, a member of IL-1 family was the first discovered interleukins, and exert proinflammatory responses through signaling via IL-1R1 [18]. Neuroinflammation is defined as the brain's innate immune response to injury. The hallmarks of a neuroinflammatory response are phenotypic glial activation and de novo production of immune signaling molecules.
IL-1β is intimately involved in elaboration of acute neuroinflammatory processes in vivo. Exposure of the rodent brain to IL-1β elicits rapid, robust activation of both astrocytes and microglia. Importantly, IL-1β is capable of triggering further increases in its own expression as evidenced by murine IL-1β induction following human IL-1β administration or expression in the brain [19]. By feeding back upon itself, small localized elevations in IL-1 may be sufficient to drive potent neuroinflammatory changes in the brain.
IFNγ, a pro-inflammatory cytokine, is not only produced by a restricted set of peripheral cells, such as T lymphocytes, natural killer (NK) and NKT cells, but also by central nervous system (CNS) cells in response to specific stimuli. Because T, NK and NKT cells are scarce in healthy brain parenchyma, IFNγ is thought to only play a role in brain function under pathological conditions such as CNS infections, inflammatory diseases, trauma and stroke [20]. More recently, Sun L et al. have reported that anti-IFN-γ antibodies exacerbate the acute course of neuroinflammation and lead to neurocyte apoptosis in vivo. The pro-inflammatory cytokine IFN-γ provided neuroprotection during acute neuroinflammation via induction of astrocyte-secreted IL-6 [21].
TNF, also called TNF-α, is a potent mediator of inflammation, as well as many normal physiological functions in homeostasis and health and antimicrobial immunity. neuroinflammation refers to inflammation associated with the brain and is characterized by the activation of microglia and expression of major inflammatory mediators without typical features of peripheral inflammation such as edema and neutrophil infiltration [22].
NFκB is one of the most important transcription factors that activate transcription of many proinflammatory genes, and the unraveling of TNF-α induced NFκB activation forms the foundation of TNF-α as major cytokine of neuroinflammation. It is well documented that TNF-α induces at least five different types of signals that include activation of NFκB, apoptosis pathways, extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (p38MAPK), and c-Jun N-terminal kinase (JNK) [23] [24].
MS is the prototypical inflammatory disease of the central nervous system (CNS). MS lesions harbor different immune cells, but the contribution of individual cell types to disease etiology. In experimental autoimmune encephalomyelitis (EAE), auto-reactive helper T (Th) cells instigate CNS inflammation by acting on myeloid cells via the production of granulocyte-macrophage colony-stimulating factor (GM-CSF) [25]. Moreover, GM‑CSF is the communication conduit between T H cells and myeloid cells. GM-CSF is the cytokines produced by pathogenic T H cells in response to IL-23R engagement. Although historically the discovery that GM-CSF deficiency results in EAE resistance IL7 preceded the studies regarding the role of IL-17 in neuroinflammation, only recently has the connection between T cell-derived GM-CSF and disease progression been made.
References:
[1] WEI-WEI CHEN, XIA ZHANG, et al. Role of neuroinflammation in neurodegenerative diseases (Review) [J]. Mol Med Rep. 2016, 13(4): 3391–3396.
[2] Allison DJ, Ditor DS. The common inflammatory etiology of depression and cognitive impairment: A therapeutic target [J]. J Neuroinflammation. 2014, 11:151.
[3] de Leeuw FE, de Groot JC, et al. Hypertension and cerebral white matter lesions in a prospective cohort study [J]. Brain. 2002, 125:765–772.
[4] Schiffrin EL. Inflammation, immunity and development of essential hypertension [J]. J Hypertens. 2014, 32:228–229.
[5] Shimizu M, Ishikawa J, et al. The relationship between the morning blood pressure surge and low-grade inflammation on silent cerebral infarct and clinical stroke events [J]. Atherosclerosis. 2011, 219:316–321.
[6] Candice M. Brown, Tara A. Mulcahey, et al. Production of Proinflammatory Cytokines and Chemokines During Neuroinflammation: Novel Roles for Estrogen Receptors α and β [J]. Endocrinology. 2010, 151(10): 4916–4925.
[7] Hu, W. T. et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease [J]. Neurology. 2012, 79, 897–905.
[8] Patel, N. S. et al. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease [J]. J. Neuroinflamm. 2005, 2, 9.
[9] Burkhard Becher, Sabine Spath, et al. Cytokine networks in neuroinflammation [J]. Nat Rev Immunol. 2017, 17(1):49-59.
[10] Aggarwal, S., Ghilardi, N., et al. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17 [J]. J. Biol. Chem. 2003, 278, 1910–1914.
[11] Hirokazu Hara, Dai Kimoto, et al. Apomorphine prevents LPS-induced IL-23 p19 mRNA expression via inhibition of JNK and ATF4 in HAPI cells [J]. European Journal of Pharmacology. 2017, 795: 108–114.
[12] Cua, D.J., Sherlock, J., et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain [J]. Nature. 2003, 421, 744–748.
[13] YuhongYang, Amy E.Lovett-Racke, et al. Role of IL-12/IL-23 in the Pathogenesis of Multiple Sclerosis [J]. Neuroinflammation. 2011, 6: 107-136.
[14] Li, Y., Chu, N., et al. Increased IL-23p19 expression in multiple sclerosis lesions and its induction in microglia [J]. Brain, 2007, 130, 490–501.
[15] Ya-Ying Wu, Jung-Lung Hsu, et al. Alterations of the Neuroinflammatory Markers IL-6 and TRAIL in Alzheimer's Disease [J]. Dement Geriatr Cogn Dis Extra. 2015, 5(3): 424–434.
[16] Lee KS, et al. Peripheral cytokines and chemokines in Alzheimer's disease [J]. Dement Geriatr Cogn Disord. 200, 28:281–287.
[17] Walsh KP, et al. Amyloid-beta and proinflammatory cytokines utilize a prion protein-dependent pathway to activate NADPH oxidase and induce cofilin-actin rods in hippocampal neurons [J]. PLoS One. 2014, 9:e95995.
[18] Andrew S. Mendiola and Astrid E. Cardona. The IL-1β phenomena in neuroinflammatory diseases [J]. J Neural Transm (Vienna). 2018, 125(5): 781–795.
[19] Shaftel SS, Kyrkanides S, et al. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology [J]. J Clin Invest. 2007, 117 (6): 1595-1604.
[20] S. Monteiro, S. Roque, et al. Brain interference: Revisiting the role of IFNg in the central nervous system [J]. Progress in Neurobiology. 2017.
[21] Sun L, Li Y, et al. Neuroprotection by IFN-γ via astrocyte-secreted IL-6 in acute neuroinflammation [J]. Oncotarget. 2017, 8(25):40065-40078.
[22] Song JH, Lee JW, et al. Glycyrrhizin alleviates neuroinflammation and memory deficit induced by systemic lipopolysaccharide treatment in mice [J]. Molecules. 2013, 18:15788-15803.
[23] Aggarwal BB, Gupta SC, et al. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey [J]. Blood. 2012, 119(3):651-665.
[24] Mubarak Muhammad. Tumor Necrosis Factor Alpha: A Major Cytokine of Brain Neuroinflammation. 2019.
[25] Andrew L. Croxford, Sabine Spath, et al. GM-CSF in Neuroinflammation: Licensing Myeloid Cells for Tissue Damage [J]. Trends in Immunology. 2015, 36(10): 651-662.
What are Cytokines?
Cytokines and Diseases
Related Pathways
Related Articles