Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Rheumatoid Arthrts (RA) is a classic autoimmune disease with a prevalence of 1% worldwide. If 70% of the patients are not timely diagnosed and treated, they may become disabled after 2 years, and the average life expectancy will be shortened by 10-15 years. As a result, RA has been a research hotspot all over the world.
In recent years, due to the rapid development of molecular biology and immunology, the pathogenesis and clinical treatment of RA has achieved a profound development. Recently, immune cells and various cytokines are the research focus of the mechanism of RA. Cytokines are important molecules involved in the immune inflammatory response of RA, which not only provides a deeper theoretical understanding of the occurrence of RA, but also lays a solid foundation for further clinical immunotherapy. This review pays attention on the cytokines promising in the RA therapy. The review includes following sections:
Rheumatoid arthritis (RA) is an autoimmune disease, which means that your immune system attacks healthy cells in your body by mistake, causing inflammation in the affected parts of the body. RA is a chronic disease characterized by symptoms of inflammation and pain in the joints. The joint symptoms of RA usually include joint pain, joint swelling, joint stiffness and loss of joint function and deformities. Symptoms can vary from mild to severe. It's important not to ignore your symptoms, even if they come and go. Knowing the early signs of RA will help you and your healthcare provider better treat and manage it. RA can also affect other tissues throughout the body and cause problems in organs, involving the lungs, heart, and eyes.
RA is usually diagnosed by reviewing symptoms, conducting a physical examination, and doing X-rays and lab tests. It's best to diagnose RA early—within 6 months of the onset of symptoms—so that people with the disease can begin treatment to slow or stop disease progression. Diagnosis and effective treatments, particularly treatment to suppress or control inflammation, can help mitigate the damaging effects of RA.
RA is a chronic, inflammatory joint disease of autoimmune nature. It is pathologically heterogeneous and characterized by autoantibodies to immunoglobulin G (IgG) and citrullinated proteins (ACPAs) [1]. If insufficiently treated, RA can lead to accumulating joint damage and irreversible disability [2]. This is most likely due to formation of immune complexes by anti-citrullinated protein antibodies (ACPAs) with citrullinecontaining antigens and subsequent binding of rheumatoid factor (RF), which can lead to abundant complement activation.
ACPAs can bind citrullinated residues on many self-proteins including vimentin, α-enolase, fibronectin, fibrinogen, histones, and type II collagen. The tissue in which these immune responses are activated is uncertain, but the lung is an attractive candidate, which is consistent with a role for smoking in rheumatoid arthritis and the presence of shared citrullinated peptides in lung and synovial tissue biopsies (figure 1) [3] [4].
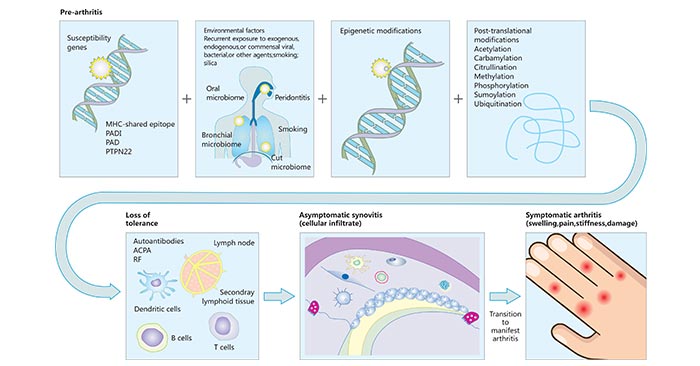
Figure 1. : Pathways to rheumatoid arthritis
*This diagram is derived from reference 1.
As you know, cytokines are cell molecules that are secreted by immune cells and aid cell to cell communication in immune responses and stimulate the movement of cells towards sites of inflammation, infection and trauma. In RA, cytokines may be classified into four groups: pro-inflammatory cytokines (IL-1 and TNFα), inflammatory cytokines in joints (IL-1, TNFα, IL-6, IL-15, IL-16, IL-17, IL-18, IFN-γ, GM-CSF), anti-inflammatory cytokines (IL-4, IL-10, IL-11, IL-13 and IL-20) and natural cytokine antagonists (IL-1ra and sTNF-RI) [5] [6] [7] [8]. As mentioned before, this review only pays attention on the cytokines promising in the RA therapy, including IL-1, IL-6, IL-17, IL-23, VEGF and TNF-α.
IL-1 in joint tissues is mainly produced by chondrocytes of synovial tiusses. IL-1 is one of the most important proinflammatory cytokines to destruct RA articular cartilage. IL-1 can stimulate the expression of PLA2 and cyclooxygenase-2 in human synovial cell, and increase their activity. Then decomposing lecithin membrane produces arachidonic acid, leading to the generation and release of PGE2 [9]. IL-1 can promote the synthesis and release of prostaglandins (PGE2) and collagenase in synovial cells and cartilage cells. IL-1 can induce synovial cell of RA joint proliferation and stimulate synovial cells to produce protein kinase.
PGE2 and collagenol trigger synovium inflammation and collapse of cartilage matrix, while decomposition products such as local immune complex and free collagen stimulate IL-1 secretion, aggravating the development of RA. Protein kinase directly acts on cartilage matrix to cause cartilage absorption, and IL-1 can also lead to the synthesis and secretion of denatured protease, causing interstitial degradation and bone destruction. IL-1 stimulates synovium and cartilage to synthesize excessive metalloproteinase-collagenol and matrix lysin. Lysin matrix can be directly destroy cartilage matrix decomposition, also can promote produce proteoglycan [10].
IL-6 is an inflammatory cytokine that has wide-ranging effects. It exerts influence on both adaptive and innate immune cells. Furthermore, it plays a role in osteoclast differentiation that may lead to bone destruction as well as mediates anemia due to chronic inflammation. In RA, it is believed that the dysregulation of IL-6 expression is an important part of disease pathogenesis. Many studies have demonstrated that IL-6 is elevated in the serum of RA patients [11]. More intriguingly, the levels of IL-6 are often highest within the synovial fluid and synovial cells are thought to be a major source of IL-6 production in RA [12].
In the past decade, IL-6 inhibition has emerged as a potent and effective treatment option for RA [13]. The first anti-IL-6 therapy developed was tocilizumab, which is a humanized monoclonal antibody against the IL-6 receptor thereby disrupting the binding of IL-6 to its cognate receptor. Tocilizumab is capable of disrupting the binding of IL-6 to both the membrane bound as well as soluble IL-6 receptor.
IL-17, commonly referred to as IL-17A, is the founding member of the IL ‑17 family, which consists of six ligands, A–F. IL‑17 was originally thought to signal via the classic receptor IL‑17R, although further investigation demonstrated that IL‑17 stimulates a receptor complex that consists of the subunits IL‑17RA and IL ‑17RC [14] [15]. Th17 cells are the major source of IL‑17. IL‑17 influences the activity of various cell types, and consequently leads to the development of features characteristic of heumatoid arthritis including inflammation and destruction of cartilage and bone.
IL‑17 overexpression in the murine knee joint induces joint inflammation, bone erosion and cartilage proteoglycan loss. IL‑17 shares catabolic properties with IL‑1 but is less potent. Interestingly, the potency of IL‑17 can be markedly increased through synergy with the effects of TNF. IL‑17 can directly activate chondrocytes to inhibit matrix synthesis and induce enzymes that degrade cartilage matrix, and thereby has a doubly catabolic effect. In addition, il‑17 is a strong amplifier of immune‑complex‑mediated arthritis [16] [17].
As you know, IL-23, a heterodimeric cytokine, is comprised of the IL-12p40, which it shares with Il-12, and the IL-23p19 subunit. It is recognized by the IL-23 receptor, which is comprised of the IL-23A and Il-12β1 receptor. Accumulating evidence have indicated that IL-23 plays a role in the differentiation of Th17 cells and the pathogenesis of RA [18]. Animal models of inducible arthritis in which IL23 has been genetically deleted demonstrate a decrease in the number of Th17 cells as well as protection from the development of arthritis. It is believed that IL-23 dependent abrogation of Th17 differentiation is able to prevent the development of disease but does not stem disease activity once the disease process has been initiated [19].
In 1994, Fava et al. found that the expression of VEGF exists in synovium tissue of RA patients by immunohistochemistry, and the staining positive cells were mainly synovium lining cells. Most fibroblasts (FLS) and a small number of macrophages were obtained from primary cell culture of synovial tissue of patients with RA. These FLS cells secrete a lot of VEGF. Clinical studies have confirmed that the higher the VEGF value in peripheral blood of RA patients, the more involved the number of joints, and the more severe the joint lesions. Therefore, some researchers believe that the VEGF level in peripheral blood can be used as an evaluation index to reflect the degree of RA patients, as well as blood sedimentation.
In addition, many cytokines are involved in the regulation of VEGF. Both IL-1 and TNF-α can promote synovial angiogenesis by upregulating the secretion of VEGF. Related experiments confirmed that the combined use of IL-1 and TNF-αantibodies to cultured synovium cells of RA could reduce the expression of VECF by 45%, but the use of only one of the antibodies had no significant effect on blocking the expression of VECGF by synovium cells.
In RA, TNF-α was mainly secreted by patients' peripheral blood monocytes and articular synovium macrophages, and TNF-α was mostly detected in the pathological tissues of joints, and TNF was detected in the articular transit of 50% of RA patients.
TNF-α is mainly involved in three pathological processes in the development of RA. First, TNF-α can increase the expression of adhesion molecules by vascular endothelial cells, so that leukocytes in the blood can be concentrated into the joint cavity through interaction with adhesion molecules. Second, TNF-α stimulates connective tissue cells and polymorphonuclear cells to produce small molecules such as prostaglandins and act as mediators of inflammation. Third, TNF-α stimulates synovium cells and chondrocytes to reduce glycoprotein synthesis in osteoclasts, increase glycoprotein degradation, and produce collagenase and other neutral proteases to release bone calcium, leading to the destruction of bone and cartilage. In immunotherapy for RA patients, the use of TNF-α-mAb or soluble TNF receptor can significantly improve clinical symptoms.
References:
[1] Josef S Smolen, Daniel Aletaha, et al. Rheumatoid arthritis [J]. Lancet. 2016.
[2] Josef S Smolen, Daniel Aletaha, et al. Rheumatoid arthritis [J]. Nat Rev Dis Primers. 2018.
[3] Anquetil F, Clavel C, et al. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor- and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies [J]. J Immunol. 2015, 194: 3664–74.
[4] Girbal-Neuhauser E, Durieux JJ, et al. The epitopes targeted by the rheumatoid arthritis-associated antifi laggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues [J]. J Immunol. 1999, 162: 585–94.
[5] Kasama, T., Isozaki, T., et al. Clinical effects of tocilizumab on cytokines and immunological factors in patients with rheumatoid arthritis [J]. Int. Immunopharmacol. 2016, 35: 301–306.
[6] Jimenez-Boj, E. et al. Interaction between synovial inflammatory tissue and bone marrow in rheumatoid arthritis [J]. J. Immunol. 2005, 175: 2579–2588.
[7] Choy, E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis [J]. Rheumatology 2012, 51: 3–11.
[8] Ponchel, F., Goéb, V., et al. An immunological biomarker to predict MTX response in early RA [J]. Ann. Rheum. Dis. 2014, 73(11): 2047–2053.
[9] Ghezzi P, Cerami A. Tumor necrosis factor as a pharmacological target [J]. Methods Mol Med. 2004, 98 :1-8.
[10] Niki Y, Yamada H, et al. Membrane-associated IL- 1contributes to chronic svnovitis and cartilare destruction in human IL-1 alpha transgenic mice [J]. J Immunol. 2004, 72 (1) 577-584.
[11] Baumann, H. & Kushner, I. Production of interleukin-6 by synovial fibroblasts in rheumatoid arthritis [J]. Am. J. Pathol. 1998, 152, 641–644.
[12] Stone, J. H. et al. Trial of Tocilizumab in Giant-Cell Arteritis [J]. N. Engl. J. Med. 2017, 377, 317–328.
[13] Viet L. Bui, Ernie Brahn. Cytokine targeting in rheumatoid arthritis [J]. Immunol. 2019, 29: 613-621.
[14] Gaffen, S. L. An overview of IL‑17 function and signaling [j]. Cytokine. 2008, 43, 402–407.
[15] Maitra, A. et al. Distinct functional motifs within the IL‑17 receptor regulate signal transduction and target gene expression [J]. Proc. Natl Acad. Sci. USA. 2007, 104, 7507–7511.
[16] Geboes, L. et al. Proinflammatory role of the TH17 cytokine IL‑22 in collagen‑induced arthritis in C57Bl/6 mice [J]. Arthritis Rheum. 2009, 60, 390–395.
[17] Wim B. van den Berg and Pierre Miossec. Il‑17 as a future therapeutic target for rheumatoid arthritis [J]. Nat Rev Rheumatol. 2009, 5: 549-553.
[18] Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis [J]. Nature Reviews Rheumatology. 2015, 11, 415–429.
[19] Cornelissen, F. et al. IL-23 Dependent and Independent Stages of Experimental Arthritis: No Clinical Effect of Therapeutic IL-23p19 Inhibition in Collagen-induced Arthritis [J]. PLoS One. 2013, 8.
What are Cytokines?
Cytokines and Diseases
Related Pathways
Related Articles