Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
Live long enough, and most men will develop prostate cancer. Globally, it is the second most common cancer in men. Prostate cancer occurs in the prostate gland, which is located just below the bladder in males and surrounds the top portion of the tube that drains urine from the bladder. The prostate, like all other glandular organs, consists of an epithelial and a stromal compartment, which contain multiple cell types. As the Fig.1 shows, stromal and epithelial compartments in the prostate gland are separated by the basement membrane, a packed structure of collagen fibers containing various extracellular matrix proteins produced by both epithelial and stromal cells [1].
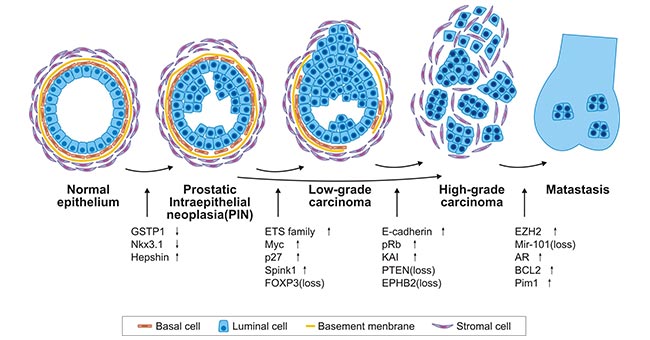
Figure 1. Model of prostate cancer progression
*this diagram is derived from publication on Adv Cancer Res [2]
In general, significant changes in both epithelial and stromal cell compartments take place during prostate cancer initiation and progression. The figure 1 depicts histological changes and concomitant genetic and epigenetic events during prostate cancer initiation and progression. The deletion or inactivating mutation in tumor-suppressor genes are denoted as (loss). Overexpression of a gene is shown with an arrow pointing up, while downregulation of expression is shown with an arrow pointing down. Note that these changes are only initial changes in the expression levels. The up or down regulation of expression may persist at more advanced stages.
In this article, we list part of targets involved in prostate cancer based on the information provided by NCG (web resource to analyze duplicability, orthology and network properties of cancer genes)
Here, we display several key targets involved in mechanism of prostate cancer, including:
PTEN (Phosphatase and tensin homolog) is a protein that acts as dual-specificity phosphatase, converting phosphatidylinositol 3,4,5-trisphosphate (PIP3) into phosphatidylinositol 4,5-bisphosphate (PIP2) [3]. Genomic aberrations of the PTEN are among the most common in prostate cancer. Inactivation of the PTEN gene by deletion or mutation is identified in ~20% of primary prostate tumor samples at radical prostatectomy and as many as 50% of castration resistant tumors. Loss of PTEN function leads to activation of the PI3K–AKT pathway and is strongly associated with adverse oncological outcomes, making PTEN a potentially useful genomic marker to distinguish indolent from aggressive disease in patients with clinically localized tumors [4].
ATM (ataxia telangiectasia mutated) encodes a PI3K-related serine/threonine protein kinase (PIKK) that helps maintain genomic integrity. ATM plays a central role in the repair of DNA double-strand breaks (DSB) [5]. And prostate cancer is associated with defective DNA strand break repair after DNA damage leading to genetic instability and prostate cancer progression [6]. Then, several scientists supposed and demonstrated that the prevalence of the ATM missense variant P1054R predisposes to prostate cancer.
SPOP (Speckle-type POZ protein) is the most commonly mutated gene in primary prostate cancer modulates DNA double strand break (DSB) repair. And SPOP mutation is associated with genomic instability. Gunther Boysen et al. has demonstrated that SPOP mutation drives prostate tumorigenesis in part through genomic instability, and indicate that mutant SPOP may increase response to DNA-damaging therapeutics [7].
APC (adenomatous polyposis coli), a key tumor suppressor gene, encodes a protein that has multiple domains, through which it binds to various proteins, including beta-catenin, axin, CtBP, Asefs, IQGAP1, EB1 and microtubules. Studies using mutant mice and cultured cells have demonstrated that APC suppresses canonical Wnt signaling, which is essential for tumorigenesis [8]. Accumulating evidence has revealed that prostate tumors induced by the deletion of Apc have elevated levels of β-catenin protein and are highly proliferative [9].
AR (Androgen receptor), a steroid receptor transcriptional factor for testosterone and dihydrotestosterone, plays pivotal roles in prostate cancer, especially castration-resistant prostate cancer (CRPC) [10]. Currently, there are eight androgen receptor antagonists approved for prostate cancer treatment, including Darolutamide, Apalutamide, Enzalutamide, Cyproterone Acetate, Bicalutamide, Flutamide, Nilutamide and Cyproterone Acetate/Ethinylestradiol.
References
[1] Bonkhoff, H., Wernert, N., Dhom, G. et al. Distribution of basement membranes in primary and metastatic carcinomas of the prostate [J]. Hum. Pathol. 1992, 23,934–939.
[2] Beatrice S. Knudsen and Valera Vasioukhin. Mechanisms of Prostate Cancer Initiation and Progression [J]. Adv Cancer Res. 2010, 109:1-50.
[3] Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate [J]. J Biol Chem. 1998, 273(22):13375-8.
[4] Tamara Jamaspishvili, David M. Berman, Ashley E. Ross et al. Clinical implications of PTEN loss in prostate cancer [J]. Nat Rev Urol. 2018, 15(4): 222–234.
[5] Michael Choi, Thomas Kipps and Razelle Kurzrock. ATM Mutations in Cancer: Therapeutic Implications [J]. Molecular Cancer Therapeutics. 2016.
[6] AndreasMeyer, BettinaWilhelm, ThiloDörk et al. ATM missense variant P1054R predisposes to prostate cancer [J]. Radiotherapy and Oncology. 2007, 83(3):283-288.
[7] Gunther Boysen, Christopher E Barbieri, Davide Prandi et al. SPOP mutation leads to genomic instability in prostate cancer [J]. Elife. 2015, 16(4): e09207.
[8] Koji Aoki, Makoto M Taketo. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene [J]. J Cell Sci. 2007, 120(Pt 19): 3327-35.
[9] Katia J. Bruxvoort, Holli M. Charbonneau, Troy A. Giambernardi, et al. Inactivation of Apc in the Mouse Prostate Causes Prostate Carcinoma [J]. Cancer Res. 2007, 67(6):2490–6.
[10] Kazutoshi Fujita , Norio Nonomura. Role of Androgen Receptor in Prostate Cancer: A Review [J]. World J Mens Health. 2019, 37(3):288-295.
Most Common Cancers
The Pathogenesis of Cancer
Related Articles
Related Pathways