Call us
301-363-4651 (Available 9 a.m. to 5 p.m. CST from Monday to Friday)
| Code | CSB-E09457h |
| Size | 96T,5×96T,10×96T |
| Price | Request a Quote |
| Trial Size |
24T ELISA Kit Trial Size (Only USD$150/ kit) * Sample kit cost can be deducted as a $30 credit for each 96-assay kit of the same analyte and brand you subsequently purchase within six months until depleted. More details >> Interested in a trial size? Please leave a message below.
|
| Have Questions? | Leave a Message or Start an on-line Chat |
| Intra-assay Precision (Precision within an assay): CV%<8% | ||||||
| Three samples of known concentration were tested twenty times on one plate to assess. | ||||||
| Inter-assay Precision (Precision between assays): CV%<10% | ||||||
| Three samples of known concentration were tested in twenty assays to assess. | ||||||
| To assess the linearity of the assay, samples were spiked with high concentrations of human αGAL in various matrices and diluted with the Sample Diluent to produce samples with values within the dynamic range of the assay. | ||||||
| Sample | Serum(n=4) | |||||
| 1:100 | Average % | 86 | ||||
| Range % | 81-92 | |||||
| 1:200 | Average % | 92 | ||||
| Range % | 87-96 | |||||
| 1:400 | Average % | 84 | ||||
| Range % | 81-86 | |||||
| 1:800 | Average % | 102 | ||||
| Range % | 100-106 | |||||
| The recovery of human αGAL spiked to levels throughout the range of the assay in various matrices was evaluated. Samples were diluted prior to assay as directed in the Sample Preparation section. | ||||||
| Sample Type | Average % Recovery | Range | ||||
| Serum (n=5) | 94 | 90-98 | ||||
| EDTA plasma (n=4) | 88 | 84-93 | ||||
| These standard curves are provided for demonstration only. A standard curve should be generated for each set of samples assayed. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
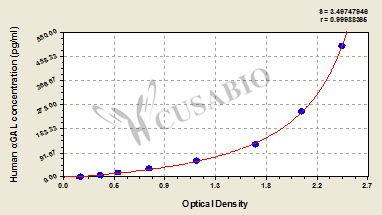
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
This Human GLA ELISA Kit was designed for the quantitative measurement of Human GLA protein in serum, plasma, tissue homogenates. It is a Sandwich ELISA kit, its detection range is 7.8 pg/mL-500 pg/mL and the sensitivity is 1.95 pg/mL.
There are currently no reviews for this product.

